migration from Cyprus, the Indian subcontinent, Southeast Asia, and the Middle East, it is estimated that 0.37 per 1,000 fetuses have a major hemoglobin disorder, 20% being thalassemias and 80% being sickle cell disease. A formal patient register was also recently established in Great Britain in 1997. At the end of 1999, 807 patients with thalassemia major were alive and residing in the United Kingdom, most of whom were of Pakistani or of Cypriot origin.26 Detailed information on the frequency of thalassemia in different world regions is available from Modell and Darlison,2 and Galanello et al.27
of the β-globin gene.56, 57, 58, 59 Like other genes, globin genes possess a series of motifs critical for their expression: the CAP site, which indicates the start of transcription; the AGT initiation codon, which is the signal for starting translation in messenger RNA (mRNA); the donor and acceptor splice sites, which are involved in the processing (splicing) of mRNA; the termination codon, which interrupts translation; and the polyadenylation signal, which is crucial for the addition of a poly (A) tail to the mRNA. The importance of these critical sequences is underscored by the fact that nucleotide substitutions that either alter them or create new similar consensus sequences in a globin gene cause abnormal mRNA processing and constitute the molecular basis for most types of thalassemia. Detailed information on positions, genotypes, and phenotypes for the known globin gene variants are available at the websites listed at the end of this chapter. Essentially the process of globin gene expression consists of the following steps: Transcription of DNA into a primary mRNA transcript; processing of the primary mRNA, involving modifications at both its 5′ (capping) and 3′ (polyadenylation) ends together with removal of the introns and joining of the exons (splicing) to produce mature mRNA, the final template for protein synthesis; and translation of mRNA in the globin protein. Transcription and RNA processing occur in the nucleus, while translation occurs in the cytoplasm [for review, see (15, 60)]. Thalassemia syndromes result from a large series of molecular defects, which alter the expression of one or more globin genes.
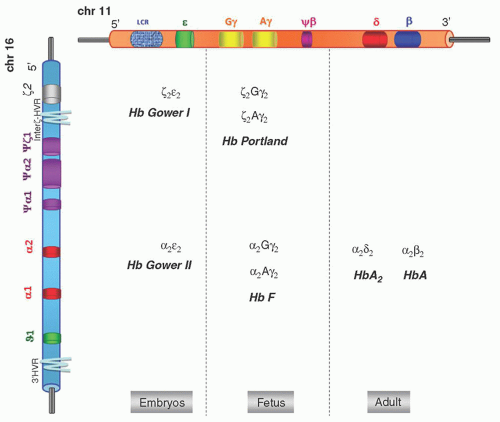 FIGURE 34.1. α– and β-globin gene cluster and hemoglobins (Hbs) produced during development. LCR, locus control region. |
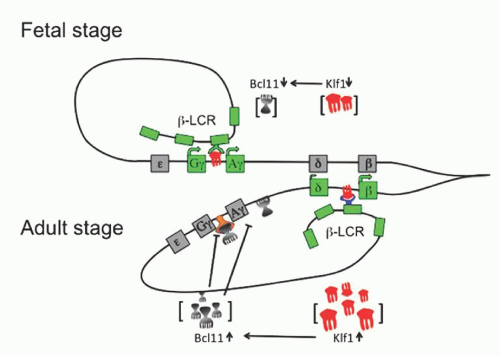 FIGURE 34.2. Current hemoglobin switching model. Bcl11A, B cell lymphoma 11; KLF, Krueppel-like factor; LCR, locus control region. (Reprinted with permission of Cao A, Moi P, Galanello R. Recent advances in β-thalassemias. Pediatr Rep 2011;3:e17.) |
α genes on each chromosome are intact (see below). The human α-globin gene cluster of apparently normal individuals contains a series of DNA sequence variations (e.g., single nucleotide polymorphism [SNP], variations in the number of tandem repeats [VNTRs]; and copy number variants [CNVs], which have been of considerable value in the analysis of evolutionary aspects of the gene cluster, in defining the origin of many of the α-thalassemia mutations, and in identifying functionally important areas of the cluster.)65, 66 The α complex is arranged in the order in which it is expressed during development: 5′ζ2 … α2–α1. There is a very high homology between α2 and α1 genes; they differ only in the IVS-2 (two base substitutions and a 7-bp insertion/deletion) and in the 3′ noncoding region (18 base substitutions and a single-base deletion in the 3′ untranslated region).67, 68 This remarkable homology has been maintained during evolution through repeated rounds of gene conversion.67, 68, 69 The embryonic ζ gene shows only 58% homology with the α genes in the coding region. The level of transcription of the two α genes differs: the α2 gene expresses two to three times more α-globin than α1.70, 71 This would imply that the globin structural variants of the α2 gene should represent about 35% of the total hemoglobin, while the α1 globin mutants should represent about 15%. However, contrasting results have been reported on this point. Shakin and Liebhaber have reported identical translation profiles of α2– and α1-mRNA, and higher percentages of α2-globin variants (24% to 40% as compared to 11% to 23% for the α1-globin variants).72 Molchanova et al. confirmed the average ratio of 2.6:1, observed for α2– and α1-mRNA, but reported an average percentage of the abnormal hemoglobin in heterozygotes with α2 mutations (23.5%) to be only slightly higher than that in heterozygotes with α1 mutations (19.7%), suggesting a less efficient translation of α2-mRNA.73 It should be pointed out that, besides the rate of transcription and efficiency of translation, other factors, such as the stability of the variant, the affinity of the variant for β-chains, and the number of active α genes, may influence the final level of the abnormal hemoglobin. The issue of different expression of the two α genes is important not only for the α-globin structural variants, but also for the pathophysiology of the deletional and nondeletional forms of α-thalassemia. Normal individuals have usually four α-globin genes, but as a result of unequal genetic exchange, some may have five or six α genes while still being phenotypically normal.74, 75, 76 Multiple arrangements with three to six ζ-like embryonic genes have also been reported.77,78
change the stop codon to one amino acid, allowing mRNA translation to continue to the next in phase stop codon located within the polyadenylation signal. The result of this class of mutation is the production of a very low amount (˜1%) of an α-chain variant elongated by 31 amino acids. It has been suggested that the reason for the reduced production of the elongated variants is the instability of the mRNA due to disruption of the 3′ untranslated region.95 Other extended α-chain variants are Hb Icaria (α 142 Lys), Koya Dora (α 142 Ser), Seal Rock (α 142 Glu), and Paksé (α 142 Tyr). Heterozygotes for α-globin elongated chains, besides the presence of a very small amount of the hemoglobin variant, have the phenotype of α-thalassemia. Mutations of α-globin genes, which result in the production of highly unstable globin variants such as Hb Quong Sze (α 125 Leu→Pro), Hb Heraklion (α 37 Pro→0) and Hb Agrinio (α 29 Leu→Pro), unable to assemble in stable tetramers and thus rapidly degraded, produce the phenotype of α-thalassemia.96 A regularly updated overview of these variants can be found at the Hb Var website. A novel mechanism for nondeletion α-thalassemia has been suggested to explain the α-thalassemia phenotype with intact α genes in some Melanesian patients.64 Among 283 single nucleotide polymorphisms (SNP) identified by resequencing of approximately 213 Kb of DNA containing the α cluster and the surrounding regions, one SNP creating a GATA-1 binding site, always linked with α-thalassemia phenotype, has been identified.97 This new GATA-1 site, which binds transcription factors in vivo and is activated in erythroid cells because it is located closer to MCS elements, competes most efficiently with the α-globin promoters, thereby causing α-thalassemia.
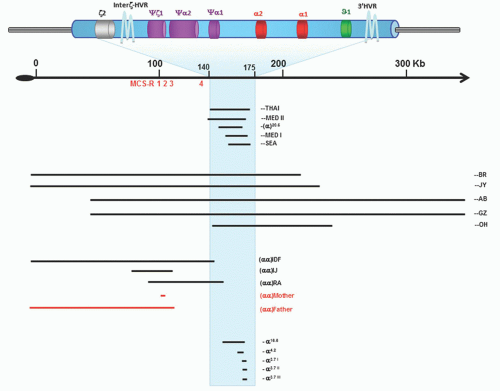 FIGURE 34.3. Most common deletional α-thalassemia defects. |
subject to a very complex regulatory mechanism, acting at the level of single genes as well as of the entire β cluster.
TABLE 34.1 MUTATIONS CAUSING β-THALASSEMIA | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
splice sites present elsewhere in precursor mRNA are used for alternative splicing, but the misspliced mRNA cannot be translated into functional β-globin.111, 112 The efficiency of normal splicing may be decreased by mutations within the consensus sequences immediately adjacent to the splice junctions. The reduction of β-globin production is quite variable and the resulting phenotypes range from mild to severe. For example, the mutations at position five of IVS-1 (G→C, G→T, G→A) produce a consistent reduction of β-globin synthesis and hence a severe β+-thalassemia phenotype; while the IVS-1-6 T→C mutation (Portuguese mutation), quite common in Mediterraneans, only mildly affects normal splicing and results in a mild thalassemia intermedia clinical picture.113 Even in the consensus sequence mutations, abnormal alternative splicing using neighboring cryptic sites may occur.112
δβ hybrid genes,
activation of cryptic splice sites,
hyperunstable β-globins, and
unknown mechanism.
and hereditary persistence of fetal hemoglobin (HPFH, see below) have not been defined. Juxtaposition to the globin genes of new sequences as a result of the deletion; removal of intergene sequences critical for control of γ-globin gene expression; and altered spatial relationships between the LCR and the genes of the β cluster (with changes in LCR/globin gene promoter interaction and competition) have been postulated to explain the upregulation of the γ-globin genes and the phenotypic differences between δβ-thalassemia and deletion HPFH. It is possible that a combination of the above mechanisms plays a role, and that a balance between regulatory sequences, with positive or negative effects on the γ gene expression, may finally determine the amount of HbF in the red cells. A recent study of three families with elevated HbF identified using comparative genomic hybridization, breakpoint DNA sequencing, and chromatin immunoprecipitation identified a 3.5 kb intergenic region near the 5′ end of the β-globin gene, which is necessary for γ-globin gene silencing.147 This region binds the fetal hemoglobin silencing factor BCL11A and its partners in the chromatin of adult erythroid cells. The Corfu δβ-thalassemia is characterized by a deletion of 7.2 kb, which removes the δ gene associated with the β-IVS-1-5 G→A mutation.148 Carriers for this mutation have the unusual hematologic phenotype of heterozygous β-thalassemia with normal levels of HbA2, while homozygotes have relatively high levels of HbF and a mild clinical phenotype.144 The 7.2-kb deletion has been reported isolated as a deletion form of δ-thalassemia not associated with increased HbF.144, 145 Two varieties of nondeletion δβ-thalassemia have also been described. One, relatively common in Sardinia, is of the δβ0 type and presents two mutations in cis: the common β039 C→T nonsense mutation, and a point mutation at position -196 in the Aγ gene promoter, which is responsible for the Italian/Chinese nondeletion Aγ HPFH (see below).149 The other, reported in two Chinese families and characterized by decreased expression of the β-globin gene and increased expression of both Gγ and Aγ globin genes (δβ+-thalassemia), showed no deletion in the β-globin cluster.150 In one of these families, the -29 A→G mutation in the promoter of the β gene (a mild β+ allele) and a non-polymorphic C→T substitution in the 3Aγ enhancer have been identified.151, 152
TABLE 34.2 MUTATIONS RESPONSIBLE FOR DELETIONAL AND NONDELETIONAL δβ-THALASSEMIA | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
phenotype of the β-thalassemia trait.166 Some mutations in the erythroid transcription factor GATA have been reported as a cause of β-thalassemia associated with thrombocytopenia.167, 168 Large somatic deletions at chromosome 11 p15.5, including the β-globin cluster and leading to thalassemia intermedia, have been reported in heterozygous β-thalassemia patients.169, 170 The deletion in a subpopulation of erythroid cells resulted in a somatic mosaic with 10% to 20% of erythroid cells heterozygous with one normal copy of the β-globin gene, and the rest homozygous without any normal β-globin gene.
TABLE 34.3 MUTATIONS RESPONSIBLE FOR HEREDITARY PERSISTENCE OF FETAL HEMOGLOBIN (HPFH) | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
of protein band 3, creating a neoantigen, which is subjected to opsonization with autologous immunoglobulin G (IgG) and complement and immune removal of the cell by macrophages.180, 181 Red blood cells in β-thalassemia lose K+, store Ca2+, and are dehydrated, resulting in altered deformability.174 Besides oxidation, free α-chains are subjected to degradation, resulting in the formation of denatured α-globin protein, heme, and free iron. These degradation products play a role in damaging erythroid precursors and red cell membranes.
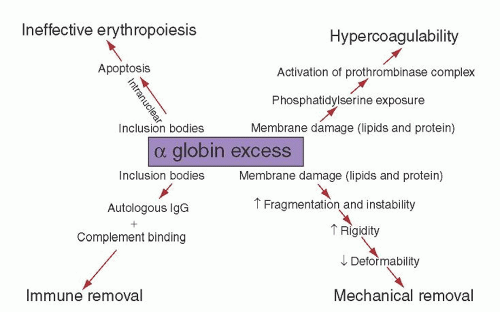 FIGURE 34.4. Pathophysiology of β-thalassemia. Ig, immunoglobulin. |
as reported in vivo in patients with HbH disease.209, 210 Interaction of excess β-globin with the cytoplasmic domain of protein band 3 is abnormal in erythrocytes of patients with HbH disease, since β4 tetramers tend to adhere tightly to protein band 3.211 β-Globin tetramers precipitate as the red cell becomes old, forming inclusions. These inclusions can be induced in vitro by vital stains, such as brilliant cresyl blue or new methylene blue, and are more common in splenectomized patients.212 Studies using monoclonal antibodies have shown that red cell inclusions in HbH disease are composed of β-globin. Membrane-bound inclusion bodies perturb the flow velocity during transit through the spleen capillaries, ultimately resulting in mechanical trapping and macrophagic phagocytosis.213 Red cell hemolysis is a significant pathophysiologic mechanism of HbH disease, but ineffective erythropoiesis is a component, even if moderate. This has been suggested by morphologic and ferrokinetic studies and by the analysis of plasma levels of the transferrin receptor.214, 215 Excess β-chains accumulate and precipitate not only in old red cells, but also in marrow erythroid precursors, where they may cause some intramedullary cell death. β4 inclusion bodies alter the normal membrane phospholipid bilayer, exposing phosphatidylserine, which represents a signal for the development of apoptosis and red cell removal by the macrophages in the spleen and other reticuloendothelial organs.216, 217 Programmed cell death has been found moderately increased in patients with HbH disease. Red cell membrane deformability and stability is even more affected in patients with HbH/Hbcs.218 As compared with patients with HbH disease, HbH/Hbcs patients have a higher amount of HbH and a higher percentage of erythrocytes with inclusion bodies and with translocated phosphatidylserine.213, 217, 219 All these characteristics may account for the increased hematologic severity of HbH/Hbcs disease. γ-Globin tetramers (Hb Bart) are much less prone than β-globin tetramers to precipitate and form inclusions.220 Besides the characteristics of excess β– and γ-globin chains discussed above, other functionally abnormal properties are important in determining the pathophysiology of α-thalassemia. HbH and, even more, Hb Bart, have a very high oxygen affinity and show no heme/heme interaction or Bohr effect, hence severely reducing their oxygen-carrying capacity.221, 222 Some α-globin chain variants barely symptomatic in the heterozygous state, are either unstable because of folding defects and/or defective in binding to α-hemoglobin-stabilizing protein (AHSP) and are associated with the α-thalassemia phenotype.96 When the patients are homozygous for such variants or compound heterozygotes with another α thalassemic defect, they present the phenotype of HbH disease or chronic hemolytic anemia or even hydrops fetalis syndrome.
present.234, 235 Studies that have correlated hematologic and clinical findings with α-globin genotypes indicate that HbH patients with nondeletion α-thalassemia defects have a more severe clinical expression (see below).236, 237, 238, 239
rather than HbH.15 The severity of HbH disease shows a good correlation with the degree of α-chain deficiency. Thus, the more severe and variable phenotypes are associated with interactions involving nondeletion α-thalassemia defects that affect the dominant α2 gene, including (-/αConstant Springα), (-/αNcoIα) and (-/αHphIα).237, 238, 239 Patients with the nondeletional genotypes present earlier, with more severe hemolytic anemia, significant growth delay, dysmorphic facial features, and more marked hepatosplenomegaly; they require more transfusions. Few patients with HbH disease resulting from the interaction of α+-thalassemia (-α) with the deletion of the MCS regulatory region have been reported.66 A severe case of HbH disease due to deletions of variable extent of both upstream MCS-Rs while all four downstream α-globin genes are intact has been recently described.92
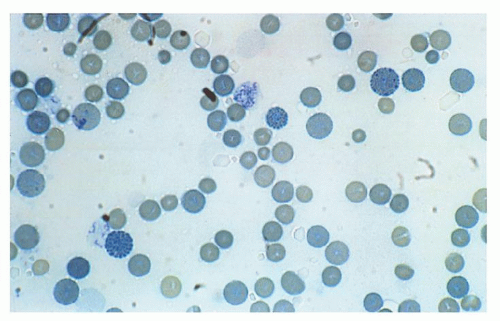 FIGURE 34.5. Hemoglobin H inclusion bodies. |
skeletal abnormalities. These subjects have extended (1 to 2 megabases) deletions resulting from rearrangements of the short arm of chromosome 16. The deletions remove both α-globin genes and up to 52 other genes.278 Two common patterns of α-thalassemia have been described: One is characterized by parents with a normal α-globin genotype (αα/αα) whose affected offspring have the phenotype of severe α-thalassemia trait (genotype -/αα). In the other, one parent has the phenotype of the mild thalassemia trait and the child has HbH disease. This condition is called ATR-16 syndrome (OMIM catalog #141750).
quartz lamp, cod liver oil, and, of course, iron) and even blood transfusions, which were helpful but short-lasting in one patient but caused increased hemolysis in another.28 This is not surprising, considering the very limited blood matching available at the time and the already enormous size of the spleen at presentation. Splenectomy and Roentgen irradiation of the spleen were also performed without benefit. All the children died shortly after presentation. Detailed autoptic data, showing peculiar abnormalities in the bones and spleen fibrosis, are available. Almost at the same time, Rietti, also from Ferrara, had reported three adult patients, two of whom were father and son, who presented with “primitive hemolytic jaundice” associated with decreased osmotic fragility.297 Anemia, microcytosis, anisocytosis, and basophilic stippling were noted. The syndrome was probably a form of thalassemia intermedia, and for a long time the eponymic title of Rietti-Greppi-Micheli was used in Italy, from the names of those who in those years described similar clinical pictures. In 1932, in consideration of the Mediterranean origin of the patients affected by Cooley anemia, Whipple and Bradford proposed the name of thalassemia, from the Greek word thalassa, meaning sea.298 Subsequently, the severe and the mild form of thalassemia were denominated thalassemia major and minor, respectively.299
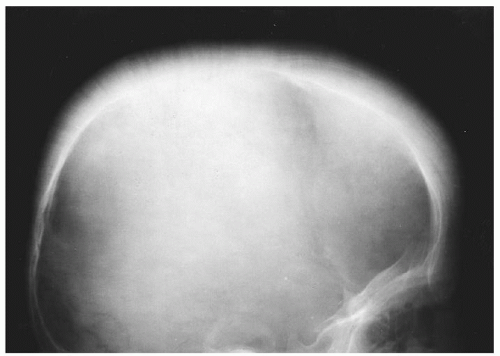 FIGURE 34.6. Radiograph of the skull. In the frontal area, the bone has a lamellated structure, parallel to the inner table of the diploe. In the parietal area, erythroid hyperplasia has perforated the outer table, producing a characteristic “hair on end” appearance. (Courtesy of Dr. C. Orzincolo.) |
≥2.5 SD below the young adult mean value, while a decrease between -1 and -2.5 SD is defined as osteopenia.320 Osteoporosis in thalassemia has been found to affect 48% of the patients, with an additional 44% affected by osteopenia.321 Although more frequent and severe in males than in females, this complication represents an important cause of morbidity in adult patients of both sexes.321 The pathogenesis of osteoporosis in thalassemia major is multifactorial and results from a variety of genetic and acquired factors. The polymorphism at the Sp1 site of the collagen type I gene (COLIA 1) has been associated with severe osteoporosis and pathologic fractures of the spine and the hip.321, 322, 323 Moreover, the vitamin D receptor (VDR) Bsm1 and Fok1 polymorphisms were found to be risk factors for bone mineral damage, low bone mineral density, and short stature in prepubertal and pubertal patients.324 However, different studies of genetic polymorphisms have given contradictory results.322, 325 Acquired factors include the primary disease itself, causing ineffective hematopoiesis with progressive bone marrow expansion; and several secondary factors such as endocrine dysfunction, iron overload and chelation therapy, vitamin deficiencies, and decreased physical activity.326, 327 In particular, vitamin D deficiency is frequent among adolescents.328 Male sex, lack of spontaneous puberty, and diabetes represent significant risk factors for osteoporosis, while transfusional history, chelation, and erythropoietic activity do not.329
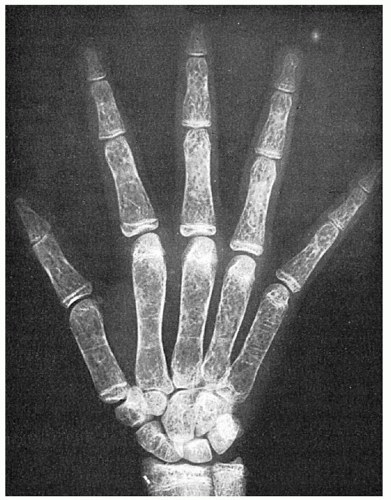 FIGURE 34.7. Mosaic pattern produced by trabeculation in the bones of the hand of a patient with thalassemia major. Note the rectangular contour of the metacarpals. |
frequency of thromboembolic events was found to be 4% in patients with thalassemia major and 10% in patients with thalassemia intermedia.352 Other groups have reported similar prevalences.353 In a large study of 8860 patients from different countries, female sex, previous splenectomy, and profound anemia were found to represent risk factors.354 A chronic hypercoagulable state has been observed even in childhood.205 The mechanisms underlying hypercoagulability in thalassemia are still unclear. Several authors197, 200 have suggested that the presence of a chronic hypercoagulable state could be due to the procoagulant effect of the anionic phospholipids exposed on the surface of the damaged circulating red blood cells and to endothelial derangement occurring as a consequence of an inflammatory state associated with the disease. Also, blood cells and platelets have an important role in increasing the thrombotic risk in thalassemia.355
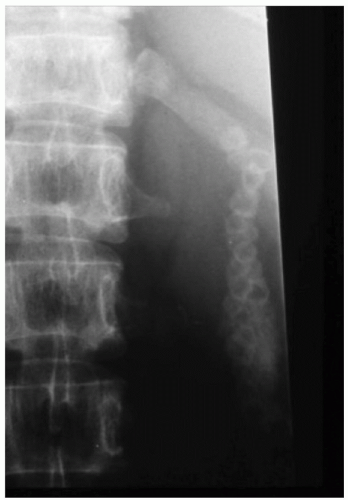 FIGURE 34.8. Spine of a 35-year-old patient with thalassemia major. Reduced mineral bone density is evident, in addition to a gallstone-filled gallbladder. |
an early biomarker of the mineralization process.378 The mechanisms by which magnesium may prevent calcium phosphate deposition in tissues are not clear, but a marked increase in the urinary output of calcium, with concomitant reduction in phosphate, has been obtained in these mice.378, 379
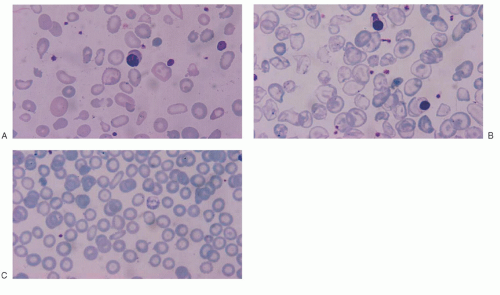 FIGURE 34.9. Peripheral blood smears in β-thalassemia major (A) and intermedia (B) and in heterozygous β-thalassemia (C). |
of both extramedullary erythropoiesis and hemolytic activity in the reticuloendothelial tissue. The bone deformities caused by the expanded marrow are typical of thalassemia and gave to all the poorly transfused patients similar features (see above). In the 1960s, however, the superiority of regular and methodically repeated transfusions was recognized, first by Orsini in France, and later by Wolman in Philadelphia and Piomelli in New York, who started a program of chronic transfusion directed at maintaining a baseline Hb level adequate to eliminate hypoxia and thus suppress its consequences.387, 388, 389 It was calculated then that the amount of iron administered to maintain a minimum Hb of 9.5 g/dl was only 50% greater than that resulting from a baseline Hb of 6 g/dl, and that the additional iron intake could be counterbalanced in part by the reduction of intestinal iron absorption. This kind of regimen, that never allowed Hb level to fall below 9.5 to 10 g/dl, was termed “hypertransfusion.” Complete bone marrow suppression, however, is seldom obtained at these Hb levels, and therefore some bone remodeling and expansion of the blood volume persist. To completely correct the effects of anemia, Propper et al. in 1980 launched what was called a “supertransfusion” regimen, where the pretransfusion hematocrit was kept at ≥ 35%.390 The hypothesis was that, as a consequence of the reduction of the blood volume, the amount of blood needed to maintain a higher baseline would not have been greater than the blood volume used for the lower baseline. A few papers from Europe confirmed the data; but, since the blood that is destroyed between transfusions and that needs replacing is a percentage of the patient’s red cell mass, patients kept at a higher baseline Hb level require a larger amount of blood and therefore accumulate more iron.391, 392, 393 In a study of patients kept at a pretransfusion hemoglobin level between 9 and 10 g/dl, the erythroid marrow activity, evaluated through the measurement of serum transferrin receptor, did not exceed two to three times normal levels. On the basis of these studies, the majority of centers choose to transfuse at a Hb level of 9 to 10.5 g/dl.394 The recommended post transfusion Hb is 14 to 15 g/dl. Leuko-reduced packed red cells are recommended for eliminating the adverse reactions attributed to contaminating white cells and for preventing platelet alloimmunization. The number of residual leukocytes should not be higher than 1 × 106. At present the preferred method for leukoreduction is prestorage filtration of whole blood with an inline filter within 8 hours after blood collection. Alternatively, laboratory filtration can be used pretransfusion. With this method, packed red blood cells are prepared from donor whole blood, then filtered prior to release from the blood bank. Finally, the packed red cell unit can be filtered at the bedside.
Asia, whereas HCV chronic infection is widespread throughout the world. The DNA-recombinant vaccine against hepatitis B virus, safe and effective, is available and should be administered to all patients who have not yet been infected. Hepatitis G virus and GB virus C (GBV-C) are RNA viruses that were independently identified in 1995, and were subsequently found to be two isolates of the same virus. Together with transfusion-transmitted (TT) virus, they are common among thalassemia patients but have not been found to contribute to chronic hepatocellular damage.410 West Nile Virus infection has become of concern in recent years. Epidemics have been reported, and the virus can be transmitted through blood transfusion.411, 412 In the United States, testing for West Nile virus antibodies has been implemented in 2003,413 and nucleic acid testing is widely used in Europe in endemic areas.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


