APPROVED KINASE INHIBITORS
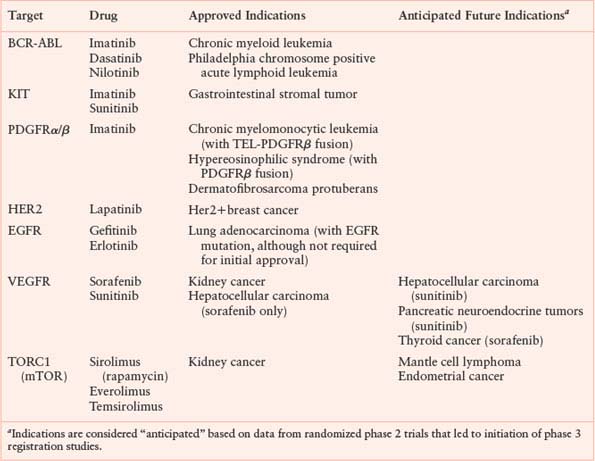
After CML, the next example to illustrate this principle was gastrointestinal stromal tumor (GIST), which is associated with mutations in the KIT tyrosine kinase receptor or, more rarely, in the platelet-derived growth factor receptor (PDGFR).4,5 Serendipitously, imatinib inhibits both KIT and PDGFR; therefore, the clinical test of KIT inhibition in GIST followed quickly on the heels of the success in CML.6 In retrospect, the rapid progress made in these two diseases was based, in part, on the fact that the driver molecular lesion (BCR-ABL or KIT mutation, respectively) is present in nearly all patients who are diagnosed with these two diseases. The molecular analysis merely confirmed the diagnosis that was made using standard clinical and histological criteria. Consequently, clinicians could identify the patients most likely to respond based on clinical criteria rather than rely on an elaborate molecular profiling infrastructure to prescreen patients. Consequently, clinical trials evaluating kinase inhibitors in CML and GIST accrued quickly and the therapeutic benefit became clear almost immediately.
The notion that molecular alteration of a driver kinase determines sensitivity to a cognate kinase inhibitor was further validated during the development of the dual EGFR/HER2 kinase inhibitor lapatinib. Clinical trials of this kinase inhibitor were conducted in women with advanced HER2-positive breast cancer based on earlier success in these same patients with the monoclonal antibody trastuzumab that targets the extracellular domain of the HER2 kinase. Lapatinib was initially approved in combination with the cytotoxic agent capecitabine for women with resistance to trastuzumab,7 then was subsequently approved for frontline use in metastatic breast cancer in combination with chemotherapy or hormonal therapy, depending on estrogen-receptor status. A key ingredient that enabled the clinical development of lapatinib was the routine use of HER2 gene amplification testing in the diagnosis of breast cancer, pioneered during the development of trastuzumab several years earlier. This widespread clinical practice allowed rapid identification of those patients most likely to benefit. If lapatinib trials had been conducted in unselected patients, the clinical signal in breast cancer would likely have been missed.
TABLE 13.2
UNAPPROVED KINASE INHIBITORS WITH CLEAR EVIDENCE OF CLINICAL ACTIVITY
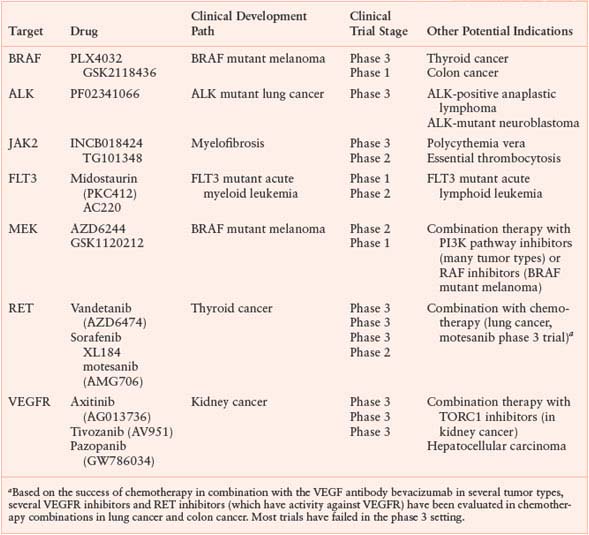
The Serendipity of Unexpected Clinical Responses
In contrast to the logical development of imatinib and lapatinib in molecularly defined patient populations, the EGFR kinase inhibitors gefitinib and erlotinib entered the clinic without the benefit of such a focused clinical development plan. Although considerable preclinical data implicated EGFR as a cancer drug target, there was little insight into which patients were most likely to benefit. The first clue that EGFR inhibitors would have a role in lung cancer came from the recognition, by several astute clinicians, of remarkable responses in a small fraction of patients with lung adenocarcinoma.8 Further studies revealed the curious clinical circumstance that those patients most likely to benefit tended to be never smokers, women, and those of Asian ethnicity.9 Clearly there was a strong clinical signal in a subgroup of patients who could perhaps be enriched based on these clinical features, but it seemed that a unifying molecular lesion must be present. Three academic groups simultaneously converged on the answer. Mutations in the EGFR gene were detected in the 10% to 15% of patients with lung adenocarcinoma who had radiographic responses.10–12 It may seem surprising that mutations in a gene as highly visible as EGFR and in such a prevalent cancer had not been detected earlier. But the motivation to search aggressively for EGFR mutations was not there until the clinical responses were seen. Perhaps even more surprising was the failure of the pharmaceutical company sponsors of the two most advanced compounds, gefitinib and erlotinib, to embrace this important discovery and refocus future clinical development plans on patients with EGFR mutant lung adenocarcinoma.
Clinical development of cytotoxic agents has always proceeded empirically. Typically, small numbers of patients with different cancers are treated in “all comer” phase 1 studies (no enrichment for subgroups) with the goal of eliciting a clinical signal in at least one tumor type. A single agent response rate of 20% to 30% in a disease-specific phase 2 trial can justify a randomized phase 3 registration trial where the typical end point for drug approval is time to progression or survival. Cytotoxics are also typically evaluated in combination with existing standard of care treatment (typically approved chemotherapy agents) with the goal of increasing the response rate or enhancing the duration of response.
The clinical development of gefitinib and erlotinib followed the cytotoxic model. Both drugs had similarly low but convincing single agent response rates (10% to 15%) in chemotherapy-refractory advanced lung cancer. Indeed, gefitinib was originally granted accelerated approval by the U.S. Food and Drug Administration (FDA) in 2003 based on the impressive nature of these responses, contingent on the completion of formal phase 3 studies with survival end points.13 The sponsors of both drugs therefore conducted phase 3 registration studies in patients with chemotherapy-refractory advanced stage lung cancer but without prescreening patients for EGFR mutation status. (In fairness, these trials were initiated prior to the discovery of EGFR mutations in lung cancer.) Erlotinib was approved in 2004 on the basis of a modest survival advantage over placebo (the BR.21 trial); however, gefitinib failed to demonstrate a survival advantage in essentially the same patient population.14,15 This difference in outcome was surprising since the two drugs have highly similar chemical structures and biological properties. Perhaps the most important difference in the two trials was drug dose. Erlotinib was given at the maximum tolerated dose, which produces a high frequency of rash and diarrhea. Both side effects are presumed “on target” consequences of EGFR inhibition since EGFR is highly expressed in skin and gastrointestinal epithelial cells. In contrast, gefitinib was dosed slightly lower to mitigate these toxicities, with the rationale that responses should not be compromised since clinical responses were clearly documented at lower doses.
In parallel with the single agent phase 3 trials in chemotherapy-refractory patients, both gefitinib and erlotinib were studied as upfront therapy for advanced lung cancer to determine if either would improve the efficacy of standard “doublet” (carboplatin/paclitaxel or gemcitabine/cisplatin) chemotherapy when all three drugs were given in combination. These trials, termed INTACT-1 and INTACT-2 (gefitinib with either gemcitabine/cisplatin or with carboplatin/paclitaxel) and TRIBUTE (erlotinib with carboplatin/paclitaxel) collectively enrolled over 3,000 patients.16–18 Excitement in the oncology community was high based on the clear single agent activity of both EGFR inhibitors. But both trials were spectacular failures—neither drug showed any benefit over chemotherapy alone. The emerging data on EGFR mutations in the 10% to 15% of patients who responded to single agent EGFR inhibitor therapy provided a logical explanation. Only a small fraction of patients enrolled in these phase 3 trials had a chance of benefit since the clinical signal from those whose tumors had EGFR mutations was diluted by all the patients whose tumors had no EGFR alterations, many of whom benefited from chemotherapy.
The convergence of the EGFR mutation discovery with these clinical trial results will be remembered as a remarkable time in the history of targeted cancer therapies, not just for the important role of these agents as lung cancer therapies but for the delays in studying whether EGFR genotype should drive treatment selection. Perhaps the most egregious error came from a retrospective analysis of tumors from patients treated in the BR.21 trial that concluded that EGFR mutations did not predict for a survival advantage.19 (EGFR gene amplification was associated with survival but only in univariate analysis.) This conclusion was concerning because less than 30% of patients on the trial had tissue available for EGFR mutation analysis, raising questions about the adequacy of the sample size. Furthermore, the EGFR mutation assay used by the authors was subsequently criticized because a significant number of the EGFR mutations reported in these patients were in residues not previously found by others who had sequenced thousands of tumors. Many of these mutations were suspected to be an artifact of working from formalin-fixed biopsies. (Formalin fixed, paraffin-embedded tissue samples present special challenges for nucleic acid analysis that can be avoided if the tissue is fresh frozen at the time of acquisition or can be overcome with additional controls.)
More recently, clinical investigators in Asia, where a greater fraction of lung cancers (roughly 30%) are positive for EGFR mutations, addressed the question of whether mutations predict for clinical benefit in a prospective trial. In this study known as IPASS, gefitinib was clearly superior to standard doublet chemotherapy as frontline therapy for patients with advanced EGFR mutation–positive lung adenocarcinoma.20 Conversely, EGFR mutation–negative patients fared much worse with gefitinib and benefited from chemotherapy. In addition, EGFR mutation–positive patients had a more favorable overall prognosis regardless of treatment, indicating that EGFR mutation is also a prognostic biomarker. The IPASS trial serves as a compelling example of a properly designed (and executed) biomarker-driven clinical trial. Although the rationale for this clinical development strategy had been demonstrated years earlier with BCR-ABL in leukemia, KIT in GIST, and HER2 in breast cancer, it was difficult to derail the empiric approach that had been used for decades in developing cytotoxic agents.
Platelet-Driven Growth Factor Receptor Leukemias and Sarcoma
The discovery of EGFR mutations in lung cancer (motivated by dramatic clinical responses in a subset of patients treated with EGFR kinase inhibitors) is the most visible example of the power of bedside-to-bench science, but it is not the only (or the first) such example from the kinase inhibitor era. Shortly after the approval of imatinib for CML in 2001, two case reports documented dramatic remissions in patients with hypereosinophilic syndrome (HES), a blood disorder characterized by prolonged elevation of eosinophil counts and subsequent organ dysfunction from eosinophil infiltration, when treated with imatinib.21,22 Although HES resembles myeloproliferative diseases such as chronic myeloid leukemia, the molecular pathogenesis of HES was completely unknown at the time. Reasoning that these clinical responses must be explained by inhibition of a driver kinase, a team of laboratory-based physician scientists quickly searched for mutations in the three kinases known to be inhibited by imatinib (ABL, KIT, and PDGFR). ABL and KIT were quickly excluded, but the PDGFRα gene was targeted by an interstitial deletion that fused the upstream FIP1L1 gene to PDGFRα.23 FIP1L1-PDGFRα is a constitutively active tyrosine kinase, analogous to BCR-ABL, and also inhibited by imatinib. As with EGFR-mutant lung cancer, the molecular pathophysiology of HES was discovered by dissecting the mechanism of response to the drug used to treat it.
The HES/FIP1L1-PDGFRα story serves as a nice bookend to an earlier discovery that the t(5,12) chromosome translocation, found rarely in patients with chronic myelomonocytic leukemia, creates the TEL-PDGFRβ fusion tyrosine kinase.24 Similar to HES, treatment of patients with t(5,12) translocation-positive leukemias with imatinib has also proven successful.25 A third example comes from dermatofibrosarcoma protuberans, a sarcoma characterized by a t(17,22) translocation that fuses the COL1A gene to the gene for PDGFB ligand (not the receptor). COL1A-PDGFB is oncogenic through autocrine stimulation of the normal PDGFR in these tumor cells. Patients with dermatofibrosarcoma protuberans respond to imatinib therapy because it targets the PDGFR, just one step downstream from the oncogenic lesion.26
Exploiting the New Paradigm: Searching for Other Kinase-Driven Cancers
The benefits of serendipity notwithstanding, the growing number of examples of successful kinase inhibitor therapy in tumors with mutation or amplification of the drug target begged for a more rational approach to drug discovery and development. In 2002, the list of human tumors known to have mutations in kinases was quite small. Due to advances in automated gene sequencing, it became possible to ask whether a much larger fraction of human cancers might also have such mutations through a brute force approach. To address this question comprehensively, one would have to sequence all of the kinases in the genome in hundreds of samples of each tumor type. Several early pilot studies demonstrated the potential of this approach by revealing important new targets for drug development. Perhaps the most spectacular was the discovery of mutations in the BRAF kinase in over half of patients with melanoma, as well as in a smaller fraction of colon and thyroid cancers27 Another was the discovery of mutations in the Janus-associated tyrosine kinase 2 (JAK2) in nearly all patients with polycythemia vera, as well as a significant fraction of patients with myelofibrosis and essential thrombocytosis.28–30 A third example was the identification of PIK3CA mutations in a variety of tumors, with the greatest frequencies in breast, endometrial, and colorectal cancers.31 PIK3CA encodes a lipid kinase that generates the second messenger phosphatidyl inositol 3-phosphate (PIP3). PIP3 activates growth and survival signaling through the AKT family of kinases as well as other downstream effectors. Coupled with the well-established role of the PTEN (phosphatase and tensin homologue) lipid phosphatase in dephosphorylating PIP3, the discovery of PIK3CA mutations focused tremendous attention on developing inhibitors at multiple levels of this pathway, as discussed further below.
Each of these important discoveries—BRAF, JAK2, PIK3CA—came from relatively small efforts (less than 100 tumors) generally focused on resequencing only those exons that coded for regions of kinases where mutations had been found in other kinases (typically the juxtamembrane and kinase domains). These restricted searches were largely driven by cost. In 2006, a comprehensive effort to sequence all of the exons in all kinases in 100 tumors could easily exceed several million dollars. Financial support for such projects could not be obtained easily through traditional funding agencies as the risk/reward ratio was considered too high. Furthermore, substantial infrastructure for sample acquisition, microdissection of tumor from normal tissue, nucleic acid preparation, high throughput automated sequencing, and computational analysis of the resulting data were essential. Few institutions were equipped to address these challenges. In response, the National Cancer Institute in the United States (in partnership with the National Human Genome Research Institute) and an international group known as the International Cancer Genome Consortium (ICGC) launched large-scale efforts to sequence the complete genomes of thousands of cancers. By 2010 the U.S. effort (called The Cancer Genome Atlas [TCGA]) had completed an analysis of glioblastoma32 and ovarian cancer and had launched efforts in lung, kidney, endometrial, and other cancers. The international consortium had committed to sequencing 25,000 tumors representing 50 different cancer subtypes.33 Both groups stipulated immediate release of all sequence information to the research community free of charge so that the entire scientific community could learn from the data. These grand-scale projects were possible due to a dramatic decline in sequencing costs. Discussions just a few years earlier about which genes (or regions of genes) to include in such projects were no longer necessary. The cost of complete exome (all exons from all genes in the human genome—not just kinases) and complete genome (all DNA) resequencing had fallen to about $5,000 per sample.
Rounding Out the Treatment of Myeloproliferative Disorders: JAK2 and Myelofibrosis
Taken together with the BCR-ABL translocation in chronic myeloid leukemia and FIP1L1-PDGFRα in HES, the discovery of JAK2 mutations in polycythemia, essential thrombocytosis, and myelofibrosis provided a unifying understanding of myeloproliferative disorders as diseases of abnormal kinase activation. JAK family kinases are the primary effectors of signaling through inflammatory cytokine receptors and had therefore been considered compelling targets for anti-inflammatory drugs. But the JAK2 mutation discovery immediately shifted these efforts toward developing JAK2 inhibitors for myeloproliferative disorders. Since most patients have a common JAK2 V617F mutation, these efforts could rapidly focus on screening for activity against a single genotype. Progress has been rapid. Two compounds (INCB018424 and TG101348) were active in phase 2 studies in myelofibrosis, one of which has already progressed to a randomized phase 3 trial.34,35 Myelofibrosis was selected as the initial indication (instead of essential thrombocytosis or polycythemia vera) because the time to registration is expected to be shortest. Currently there are no approved treatments for myelofibrosis and shrinkage in spleen size can be used as an end point for drug approval. Successful trials in myelofibrosis will likely lead to studies in essential thrombocytosis and polycythemia vera.
BRAF Mutant Melanoma: Several Missteps before Finding the Right Inhibitor
As with JAK2 mutations in myeloproliferative disorders, the discovery of BRAF mutations in patients with melanoma launched widespread efforts to find potent BRAF inhibitors. One early candidate was the drug sorafenib, which had been optimized during drug discovery to inhibit RAF kinases. (Sorafenib also inhibits vascular endothelial growth factor receptor [VEGFR], which led to its approval in kidney cancer as discussed later in this chapter.) Despite the compelling molecular rationale for targeting BRAF, clinical results of sorafenib in melanoma were extremely disappointing and reduced enthusiasm for pursuing BRAF as a drug target.36 In hindsight, this concern was completely misguided. Sorafenib dosing is limited by toxicities that preclude achieving serum levels in patients that potently inhibit RAF (but are sufficient to inhibit VEGFR). In addition, patients were enrolled without screening for BRAF mutations in their tumors. Although the frequency of BRAF mutations in melanoma is high, the inclusion of patients without BRAF mutation diluted the chance of seeing any clinical signal. In short, the clinical evaluation of sorafenib in melanoma was poorly designed to test the hypothesis that BRAF is a therapeutic target. The danger is that negative data from such clinical experiments can slow subsequent progress. It is critical to know the pharmacodynamic properties of the drug and the molecular phenotype of the patients being studied when interpreting the results of a negative study.
The fact that RAF kinases are intermediate components of the well-characterized RAS/MAP kinase pathway (transducing signals from RAS to RAF to MEK to ERK) raised the possibility that tumors with BRAF mutations might respond to inhibitors of one of these downstream kinases (Fig. 13.1). Preclinical studies revealed that tumor cell lines with BRAF mutation were exquisitely sensitive to inhibitors of the downstream kinase MEK.37 Sorafenib, in contrast, does not show this profile of activity.38 Thus, proper preclinical screening would have revealed the shortcomings of sorafenib as a BRAF inhibitor. Curiously, cell lines with mutation or amplification of EGFR or HER2, which function upstream in the pathway, were insensitive to MEK inhibition. Even tumor lines with RAS mutations were variably sensitive. In short, the preclinical data made a strong case that MEK inhibitors should be effective in BRAF mutant melanoma but not in other subtypes. The reason that HER2, EGFR, and RAS mutant tumors were not sensitive to MEK inhibitors is explained, at least in part, by the existence of negative feedback loops that modulate the flux of signal transduction through MEK.39
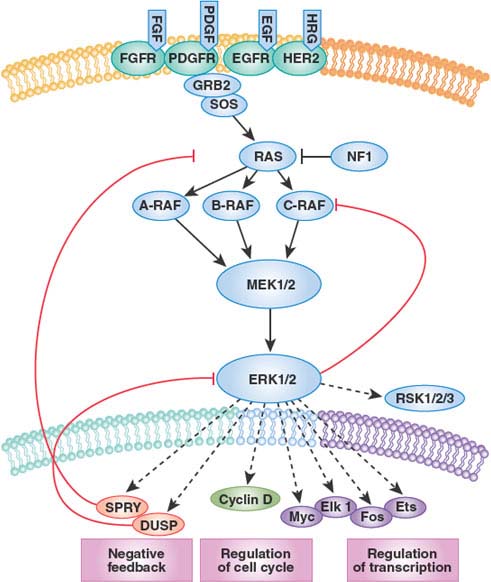
FIGURE 13.1 The RAS-RAF-MEK-ERK signaling pathway. The classical mitogen-activated protein kinase (MAPK) pathway is activated in human tumors by several mechanisms, including the binding of ligand to receptor tyrosine kinases (RTK), mutational activation of an RTK, by loss of the tumor suppressor NF1, or by mutations in RAS, BRAF, and MEK1. Phosphorylation and thus activation of ERK regulates transcription of target genes that promote cell cycle progression and tumor survival. The ERK pathway contains a classical feedback loop in which the expression of feedback elements such as SPRY and DUSP family proteins are regulated by the level of ERK activity. Loss of expression of SPRY and DUSP family members due to promoter methylation or deletion is thus permissive for persistently elevated pathway output. In the case of tumors with mutant BRAF, pathway output is enhanced by impaired upstream feedback regulation. (From ref. 114, with permission.)
In parallel with the generation of these preclinical findings, clinical trials of several MEK inhibitors were initiated. Patients with various cancers were enrolled in the early studies but there was a strong bias to include melanoma patients. Significant efforts were made to demonstrate MEK inhibition in tumor cells by measuring the phosphorylation status of the direct downstream substrate ERK using immunohistochemical analysis of biopsies from patients with metastatic disease. Phase 1 studies of the two earliest compounds in clinical development (PD325901 and AZD6244) documented reduced phospho-ERK staining at multiple dose levels in several patients on whom baseline and treatment biopsies were obtained.40,41 These pharmacodynamic studies, while well intentioned, were not quantitative enough to document the magnitude of MEK inhibition in these patients. Furthermore, clinical responses were observed in a few patients with BRAF mutant melanoma. Armed with this confidence, a randomized phase 2 clinical trial of AZD6244 was conducted in advanced melanoma, with the chemotherapeutic agent temozolomide (approved for glioblastoma) as the comparator arm. Clinical development of PD325901 was discontinued because of safety concerns about ocular and neurologic toxicity. Disappointingly, patients receiving AZD6244 had no benefit in progression-free survival when compared to temozolomide-treated patients, raising further concerns about the viability of BRAF as a drug target.42 Closer examination of the data revealed that clinical responses were, indeed, seen in patients receiving AZD6244. The fact that BRAF mutation status was not required for study entry likely diminished the clinical signal in the AZD6244 arm, a lesson learned from the EGFR inhibitor trials in lung cancer. In addition, the results in the control group were better than expected and reflect greater activity of temozolomide in melanoma than previously believed.
All doubts about BRAF as a target vanished in 2009 to 2010 when dramatic clinical responses were observed with a novel BRAF inhibitor PLX4032. Like sorafenib, this compound was optimized to inhibit RAF but with an additional focus on mutant BRAF. PLX4032 differs dramatically from sorafenib because it potently inhibits BRAF without the additional broad range of activities that sorafenib has against other kinases like VEGFR.43 The greater selectivity of PLX4032 relative to sorafenib resulted in much greater tolerability such that it could be given at high doses while avoiding significant toxicity. The early days of PLX4032 clinical development were plagued by challenges in maximizing the oral bioavailability of the drug.44 Consequently, the initial phase 1 clinical trial was temporarily halted to develop a novel formulation (the coingredients in the drug capsule or tablet that improve solubility and absorption through the gastrointestinal tract). Much higher serum levels were obtained in patients who received the new PLX4032 formation and, shortly thereafter, complete and partial responses were observed in about 80% of the melanoma patients with BRAF mutant tumors. Strikingly, no activity was observed in patients whose tumors were wild type for BRAF.45,46 The data were so compelling that PLX4032 was immediately advanced to a phase 3 registration trial. Similarly impressive responses in BRAF mutant melanoma patients were observed in a phase 1 trial of another, even more potent RAF inhibitor GSK2118436,47 providing further proof that BRAF is an important cancer target.
The PLX4032 and GSK2118436 data also provide insight into why sorafenib and the early MEK inhibitor trials failed to demonstrate activity. One lesson is the critical importance of achieving adequate target inhibition. Clinical responses with PLX4032 were observed only after the drug was reformulated to achieve substantially higher serum levels. Reductions in phospho-ERK staining (as documented by immunohistochemistry) were documented in the earlier trials but, in retrospect, the assays were not sensitive enough to distinguish between modest (approximately 50%) kinase inhibition versus more complete BRAF or MEK inhibition. Efficacy in preclinical models is significantly improved using doses that give greater than 80% inhibition, and the human trial data suggest that this degree of pathway blockade is also required for a high clinical response rate.46 Interestingly, investigators conducting a phase 1 clinical trial with a third MEK inhibitor, GSK1120212, have reported a high response rate (approximately 40%) in BRAF-mutant melanoma patients. These data appear to be substantially better than those observed with the earlier MEK inhibitors, although not as impressive as with PLX4032.48 There is speculation that this newer MEK inhibitor inhibits MEK more completely that the earlier generation compounds, but this remains to be demonstrated in patients. Collectively, these experiences illustrate the critical need for quantitative pharmacodynamic assays to measure target inhibition early in clinical development. A second lesson is the importance of genotyping all patients for mutation or amplification of the relevant drug target. Not only does this ensure that a sufficient number of patients with the biomarker of interest are included in the study, the results also provide compelling evidence early in clinical development in support (or not) of the preclinical hypothesis.
Getting It Right: ALK and Lung Cancer
The development of the ALK inhibitor crizotinib (PF02341066), which is currently under evaluation in a phase 3 registration trial in lung cancer, illustrates how an unexpected signal obtained in a small number of patients can quickly shift a program in an entirely new direction with a high probability of success. The key ingredient is this story is a familiar one—a strong molecular hypothesis backed up by clinical response data in a small number of carefully selected patients. Crizotinib emerged from a drug discovery program at Pfizer focused on finding inhibitors of the MET receptor tyrosine kinase and entered the clinic with this target as its lead indication.49 As discussed above with imatinib, essentially all kinase inhibitors have activity against other targets (so-called off target activities), which can sometimes prove to be advantageous. Off target activities are typically discovered by screening compounds against a large panel of kinases to establish profiles of relative selectivity against the intended target. Off target activity, potency, and pharmaceutical properties (bioavailability, half-life) are all factors that influence the decision of which compound to advance to clinical development. The primary off target activity of crizotinib is against the ALK tyrosine kinase.
ALK was first identified as a candidate driver oncogene in 1994 through the cloning of the t(2,5) chromosomal translocation associated with anaplastic large cell lymphoma, which creates the NPM-ALK fusion gene.50 This discovery, together with the demonstration that NPM-ALK causes lymphoma in mice, made a compelling case for ALK as a drug target in this disease. But there was limited interest in developing ALK inhibitors because this particular lymphoma subtype is rare and most commonly found in children. Companies are generally reluctant to develop drugs solely for pediatric indications because of complexities related to dose selection and additional regulatory guidelines. Efforts to streamline this development process are under way.
In 2007, another ALK fusion gene called EML4-ALK was discovered in a small fraction of patients with lung adenocarcinoma, with an estimated frequency of 1% to 5%.51 This discovery did not immediately capture the attention of drug developers, but several academic groups that had already begun testing lung cancer patients seen at their institutions for EGFR mutations simply added an EML4-ALK fusion test to the screening panel. Astute clinical investigators participating in the phase 1 trial of crizotinib, which was designed to include patients with a broad array of advanced cancers, were aware of the off target ALK activity and enrolled several lung cancer patients with EML4-ALK fusions in the study. These patients had remarkably dramatic responses.52 This serendipitous finding in a few ALK-positive patients was confirmed in a larger cohort, resulting in the initiation of a phase 3 study in ALK-positive lung cancer, just 2 years after the discovery of the EML4-ALK fusion. Crizotinib is also being evaluated in other diseases associated with genomic alterations in ALK, including large cell anaplastic lymphoma, neuroblastoma,53 and inflammatory myofibroblastic sarcoma.54
Extending the Model to RET Mutations in Thyroid Cancer
Subsets of patients with papillary or medullary thyroid cancer have activating mutations or translocations targeting the RET tyrosine kinase receptor, raising the question of whether RET inhibitors might have a role in this disease.55 Although no drugs specifically designed to inhibit RET have entered the clinic, four compounds with off target activity against RET (vandetanib, sorafenib, motesanib, and XL184) have all shown single agent activity in phase 2 thyroid cancer studies.56–60 Three are currently under evaluation in phase 3 registration trials, one of which (vandetanib) has recently reported improved progression-free survival. Because all four compounds also inhibit VEGFR, it is unclear whether the clinical benefit observed in these studies is explained by inhibition of RET, VEGFR, or both. Unlike the crizotinib trials in ALK-positive lung cancer, enrollment in these registration studies was not restricted to patients with RET mutations. In addition to the fact that thyroid cancer patients are not routinely screened for these mutations, the primary reason for including all comers in these studies is that clinical responses are observed in a larger fraction of patients than can be accounted for based on the suspected frequency of RET mutation. Responses in patients without RET mutation (if they occur) might be explained by mutations in other genes in the RAS-MAP kinase pathway such as BRAF or HRAS, which are found in a substantial fraction of patients and are typically nonoverlapping with RET alterations.55 Clearly, detailed genotype–response correlations, as demonstrated in lung cancer and melanoma, will clarify the role of these mutations in predicting response to these drugs. Thyroid cancer is also a compelling indication for the BRAF and MEK inhibitors discussed above in melanoma.
FLT3 Inhibitors in Acute Myeloid Leukemia
Shortly after the success of imatinib, the receptor tyrosine kinase FLT3 emerged as a compelling drug candidate based on the presence of activating mutations in about one-third of patients with acute myeloid leukemia (AML).61 Laboratory studies documented that FLT3 alleles bearing these mutations, which occur as internal tandem duplications (ITDs) of the juxtamembrane domain or point mutation in the kinase domain, function as driver oncogenes in mouse models, giving phenotypes analogous to BCR-ABL.62 As with RET in thyroid cancer, no compounds had been specifically optimized to target FLT3, but several drugs with off target FLT3 activity were redirected to AML. Disappointingly, the first three of the compounds tested (midostaurin, lestaurtinib, sunitinib) showed only marginal single agent activity in relapsed AML patients, even in those with FLT3 mutations.63–65 Despite the strong molecular rationale for FLT3 as a driver lesion, questions were raised about the viability of FLT3 as a drug target. Pharmacodynamic studies showed evidence of FLT3 kinase inhibition in tumor cells, but the magnitude and duration of these effects were difficult to quantify, raising the possibility of inadequate target inhibition.63 Indeed, the dose of all three compounds was limited by toxicities believed to be independent of FLT3. A more pessimistic interpretation was that FLT3, although presumably important for initiation of AML, was no longer required for tumor maintenance due to the accumulation of additional driver genomic alterations. If true, even complete FLT3 blockade with a highly selective inhibitor would be expected to fail. But this view was not supported by the fact that clinical responses were observed in the somewhat analogous situation of single agent ABL kinase inhibitor treatment of chronic myeloid leukemia in blast crisis, where BCR-ABL is just one of many additional genomic alterations that contributes to disease progression yet complete remissions are observed in many patients.
Despite this pessimism about FLT3 as a viable drug target, two drugs are now advancing toward drug registration trials. Midostaurin, one of the early compounds that showed disappointing single agent activity in relapsed AML, is being evaluated in a randomized phase 3 trial for newly diagnosed AML combined with standard induction chemotherapy. A single arm phase 2 study showed higher and more durable remission rates in FLT3 mutant patients when compared to historical controls.66 The second compound, AC220, is a next generation FLT3 inhibitor with greater potency and specificity and with impressive single agent activity in FLT3-mutant relapsed AML—precisely the population where midostaurin and others failed.67,68 It is not clear if this success is due to more potent FLT3 inhibition in patients since detailed pharmacodynamic studies are not yet available. Assuming these compounds prove successful in AML, it will be important to examine their activity in the rare cases of pediatric acute lymphoid leukemia associated with FLT3 mutation. Although testing is still inconclusive on FLT3 inhibitors, the failure of early compounds in AML is reminiscent of the failures of early RAF and MEK inhibitors in melanoma. Collectively, these examples emphasize the importance of using optimized compounds to test a molecularly based hypothesis in patients and to focus enrollment on those patients with the relevant molecular lesion.
Kidney Cancer: Targeting the Tumor and the Host
A recurring theme in this chapter is the critical role of driver kinase mutations in guiding the development of kinase inhibitors. Ironically, four kinase inhibitors have been approved for kidney cancer over the past 5 years in a tumor type with no known kinase mutations. The most common molecular alteration in kidney cancer is loss of function in the Von Hippel-Lindau (VHL) tumor suppressor gene, resulting in activation of the hypoxia inducible factor66 pathway.69 As a consequence of VHL loss, which normally targets hypoxia-induced factor (HIF) proteins for proteosomal degradation, HIF1α and HIF2α are constitutively active transcription factors that function as oncogenes through activation of an array of downstream target genes. Among these is the angiogenesis factor VEGF, which is secreted by HIF-expressing cells and promotes the development and maintenance of tumor neovasculature. HIF-mediated secretion of VEGF by tumor cells likely explains the highly vascular histopathology of clear cell renal carcinoma. All three currently approved angiogenesis inhibitors (the monoclonal antibody bevacizumab targeting VEGF and the kinase inhibitors sorafenib and sunitinib targeting its receptor VEGFR) have single agent clinical activity in clear cell carcinoma.70–72 The high specificity of bevacizumab for VEGF leaves little doubt that the activity of this drug is explained by antiangiogenic effects. In contrast, the off target activities of sorafenib and sunitinib include several kinases expressed in kidney tumor cells, stroma, and inflammatory cells (PDGFR, RAF, RET, FLT3, and others). Interestingly, the primary effect of bevacizumab in kidney cancer is disease stabilization, whereas sorafenib and sunitinib have substantial partial response rates. This raises the question of whether the superior antitumor activity of the VEGFR kinase inhibitors is due to concurrent inhibition of other kinases. However, partial responses rates with next generation VEGFR inhibitors (axitinib, pazopanib, and tivozanib), all of which have greater potency and selectivity for VEGFR, are similarly high and reinforce the importance of VEGFR as the critical target in kidney cancer.73–75 Molecular characterization of tumors from these patients should clarify the mechanism by which each of these inhibitors exerts antitumor activity, particularly if drug-resistant kinase domain mutations are found in any of the targets at relapse.
Two inhibitors of the mammalian target of rapamycin (mTOR) kinase (temsirolimus and everolimus) are also approved for advanced renal cell carcinoma.76,77 Both temsirolimus and everolimus are known as rapalogs since both are chemical derivatives of the natural product sirolimus (rapamycin). Sirolimus was approved more than 10 years ago to prevent graft rejection in transplant recipients based on its immunosuppressive properties against T cells. Sirolimus also has potent antiproliferative effects against vascular endothelial cells and, on that basis, is incorporated into drug eluting cardiac stents to prevent coronary artery restenosis following angioplasty.78 Rapalogs differ from other kinase inhibitors discussed in this chapter in that they inhibit the kinase through an allosteric mechanism rather than by targeting the mTOR kinase domain. Because rapalogs also inhibit the growth of cancer cell lines from different tissues of origin, clinical trials were initiated to study their potential role as anticancer agents in a broad range of tumor types. Based on responses in a few phase 1 patients with different tumor types (including kidney cancer), exploratory phase 2 studies were conducted in several diseases. Single agent activity of temsirolimus was observed in a phase 2 kidney cancer study79 then confirmed in a phase 3 registration trial.76 The phase 3 everolimus trial, which was initiated after temsirolimus, was noteworthy because clinical benefit was demonstrated in patients who had progressed on the VEGFR inhibitors sorafenib or sunitinib.77
In parallel with the empirical clinical development of rapalogs, various laboratories explored the molecular basis for mTOR dependence in cancer cells. mTOR functions at the center of a complex network that integrates signals from growth factor receptors and nutrient sensors to regulate cell growth and size (Fig. 13.2). It does so, in part, by controlling the translation of various mRNAs with complex 5’ untranslated regions into protein. mTOR exists in two distinct complexes known as TORC1 and TORC2. Rapalogs only inhibit the TORC1 complex, which is largely responsible for downstream phosphorylation of targets such as S6K1/2 and 4EBP1/2, which regulate protein translation.80 The TORC2 complex contributes to activation of AKT by phosphorylating the important regulatory serine residue S473 and is unaffected by rapalogs.
Two hypotheses have emerged to explain the clinical activity of rapalogs in kidney cancer. The antiproliferative activity of these compounds against endothelial cells suggests an antiangiogenic mechanism and is consistent with the clinical activity of the VEGFR inhibitors. But rapalogs also inhibit the growth of kidney cancer cell lines in laboratory models where the effects on tumor angiogenesis have been eliminated. Interestingly, mRNAs for HIF1/2 are among those whose translation is impaired by rapalogs, and this effect has been implicated as the primary mechanism of rapalog activity in kidney cancer xenograft models.81 As with the VEGFR inhibitors, detailed molecular annotation of tumors from responders and nonresponders will shed light on these issues.
Other Potential Indications for Mammalian Target of Rapamycin Inhibitors
The fact that rapalogs inhibit the growth of cancer cell lines and xenografts from different tumor types raised hopes for a broad role in cancer, but clinical trials of rapalogs outside of kidney cancer have generally been disappointing. Two exceptions are mantle cell lymphoma and endometrial cancer, where convincing single agent activity has led to ongoing drug registration trials.82–85 Mantle cell lymphoma is characterized by a chromosome translocation that results in constitutive expression of the cell cycle regulatory protein cyclin D1, which functions as a driver oncogene in this disease. Rapalogs inhibit translation of cyclin D1 mRNA in laboratory models, analogous to the inhibition of HIF1/2 translation by rapalogs observed in VHL-mutant kidney cancer models. Endometrial cancers often have mutations in one of several genes in the PI3K pathway (PTEN, PIK3CA, PIK3R1), any one of which causes pathway activation. Because mTOR functions downstream of PI3K, tumors with these mutations should, in theory, respond to rapalogs. Many studies of cell lines, xenografts, and genetically engineered mouse models have linked PI3K pathway activation to increased sensitivity to rapalogs.86,87 In fact, endometrial hyperplasia in mice with heterozygous loss of Pten is completely reversible by temsirolimus treatment.88 As with kidney cancer, molecular annotation of the tumors from the mantle cell lymphoma and endometrial cancer patients on these trials is essential to address these questions.
It is unclear why rapalogs have failed in other tumor types. One likely explanation is the concurrence of PI3K pathway mutations with alterations in other pathways that mitigate sensitivity to rapalogs. Another possibility is the disruption of negative feedback loops regulated by mTOR that inhibit signaling from upstream receptor tyrosine kinases. Rapalogs paradoxically increase signaling through PI3K due to loss of this negative feedback. A primary consequence is increased AKT activation, which signals to an array of downstream substrates that can enhance cell proliferation and survival (other than TORC1, which remains inhibited by rapalog) (Fig. 13.2). This problem might be overcome by combining rapalogs with an inhibitor of an upstream kinase in the feedback loop, such as the insulin-like growth factor receptor (IGFR), to block this undesired effect of rapalogs on PI3K activation.89 Alternatively, this feedback-mediated activation could, in theory, prevent use of inhibitors of PI3K itself or of the TORC2 complex that activates AKT.
TARGETING THE PI3K PATHWAY DIRECTLY
Mutations or copy number alterations (amplification or deletion of oncogenes or tumor suppressor genes) in PI3K pathway genes (PIK3CA, PIK3R1, PTEN, AKT1, and others) are among the most common abnormalities in cancer. Consequently, intensive efforts at many pharmaceutical companies have been devoted to the discovery of small molecule inhibitors targeting kinases in the PI3K pathway. Inhibitors of PI3K-, AKT-, and ATP-competitive (rather than allosteric) inhibitors of mTOR that target both the TORC1 and TORC2 complex are all in clinical development (Table 13.3). Phase 1 clinical trials have, in general, established that the pathway can be efficiently targeted without serious toxicity other than easily manageable effects on glucose metabolism (anticipated based on the importance of PI3K signaling in insulin signaling). Unfortunately, there has been no evidence to date of dramatic single agent clinical activity with any of these agents. To be fair, most compounds are still in a very early clinical development stage and have been evaluated without enriching for patients with PI3K pathway mutations.
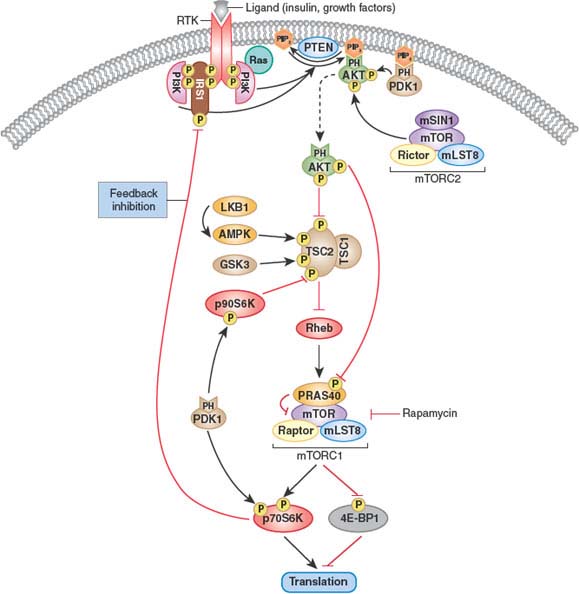
FIGURE 13.2 Feedback inhibition of the phosphatidylinositol 3-kinase (PI3K) pathway. Activated AKT regulates cellular growth through mammalian target of rapamycin (mTOR), a key player in protein synthesis and translation. mTOR forms part of two distinct complexes known as mTORC1 (which contains mTOR, Raptor, mLST8, and PRAS40) and mTORC2 (which contains mTOR, Rictor, mLST8, and mSIN1). mTORC1 is sensitive to rapamycin and controls protein synthesis and translation, at least in part, through p70S6K and eukaryotic translation initiation factor 4E–binding protein 1 (4E-BP1). AKT phosphorylates and inhibits tuberous sclerosis complex 2 (TSC2), resulting in increased mTORC1 activity. AKT also phosphorylates PRAS40, thus relieving the PRAS40 inhibitory effect on mTOR and the mTORC1 complex. mTORC2 and 3-phosphoinositide-dependent kinase (PDK1) phosphorylate AKT on Ser473 and Thr308, respectively, rendering it fully active. mTORC1-activated p70S6K can phosphorylate insulin receptor substrate 1 (IRS1), resulting in inhibition of PI3K activity. In addition, PDK1 phosphorylates and activates p70S6K and p90S6K. The latter has been shown to inhibit TSC2 activity through direct phosphorylation. Conversely, LKB1-activated AMP-activated protein kinase (AMPK) and glycogen synthase kinase 3 (GSK3) activate the TSC1/TSC2 complex through direct phosphorylation of TSC2. Thus, signals through PI3K as well as through LKB1 and AMPK converge on mTORC1. Inhibition of mTORC1 can lead to increased insulin receptor–mediated signaling, and inhibition of PDK1 may lead to activation of mTORC1 and may, paradoxically, promote tumor growth. (From ref. 115, with permission.)
TABLE 13.3
COMPOUNDS IN CLINICAL DEVELOPMENT THAT TARGET THE PI3K PATHWAY
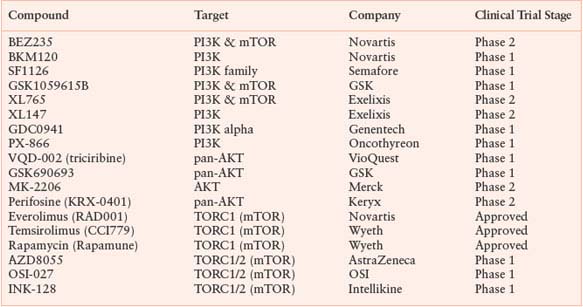
Conducting a PI3K pathway mutation-enriched trial is challenging because no single tumor type has such a high frequency of mutations that it becomes the obvious disease for early clinical development (in contrast to BRAF in melanoma). A compelling case can be made for PIK3CA mutant breast or endometrial cancer, but these trials are not straightforward because PIK3CA mutations in breast cancer are associated with good prognosis disease that is often cured by standard therapy.90 A further complication is the fact that mutations in the PI3K pathway can occur in one of many different genes in the pathway (PIK3CA, PIK3R1, PTEN, AKT1, etc.) and sometimes in combination.91 Treatment response may vary depending on the type of mutation; therefore, lumping patients with different PI3K pathway mutations into the same trial may complicate data interpretation. Indeed, tumor cell lines with PIK3CA mutation respond differently to a PI3K pathway inhibitor compared to those with PTEN loss.92 There are also technical challenges in developing an assay to determine mutation status for genes like PTEN, which can be inactivated by point mutation, deletion, or transcriptional silencing. The success or failure of these drugs as single agents should become clear in the next few years based on the outcome of many phase 2 studies that are currently under way.
COMBINATIONS OF KINASE INHIBITORS
Preclinical studies indicate that combinations of kinase inhibitors are required to realize their full potential as anticancer agents. The most common rationale is to address the problem of concurrent mutations in different pathways that alleviate dependence on a single driver oncogene. The best examples are cancers with mutations in both the RAS/MAP kinase pathway (RAS or BRAF) and the PI3K pathway (PIK3CA or PTEN). In mouse models, such doubly mutant tumors fail to respond to single agent treatment with either an AKT inhibitor or a MEK inhibitor. However, combination treatment can give dramatic regressions.93 Similarly, genetically engineered mice that develop KRAS-driven lung cancer respond only to combination therapy with a PI3K inhibitor and a MEK inhibitor.94 Several combination therapy trials combining different PI3K pathway and RAS/MAP kinase pathway inhibitors have recently been initiated.
Many of the tumor types discussed in this chapter do respond to treatment with a single agent kinase but relapse despite continued inhibitor therapy. Research into the causes of “acquired” kinase inhibitor resistance has revealed two primary mechanisms: novel mutations in the kinase domain of the drug target that preclude inhibition, or “bypass” of the driver kinase signal by activation of a parallel kinase pathway. In both cases, the solution is combination therapy to prevent the emergence of resistance. An elegant demonstration of this approach comes from CML where resistance to imatinib is primarily caused by mutations in the BCR-ABL kinase domain.95,96 The second-generation ABL inhibitors dasatinib and nilotinib are effective against most imatinib-resistant BCR-ABL mutants and were initially approved as single agent therapy for imatinib-resistant chronic myeloid leukemia.97,98 Very recently, both drugs have proven superior to imatinib in upfront treatment of chronic myeloid leukemia, due to increased potency and fewer mechanisms of acquired resistance.99–101 However, one BCR-ABL mutation called T315I is resistant to all three drugs. A novel ABL kinase inhibitor AP24534 that blocks T315I has shown promising activity in a phase 1 clinical trial that included CML patients with the T315I mutation.102,103 If AP24534 proves successful, a two drug combination of AP24534 with either dasatinib or nilotinib would likely shut off all mechanisms of relapse, analogous to triple drug highly active antiretroviral therapy (HAART) for human immunodeficiency virus (HIV). Analogous approaches should also be successful in other diseases such as EGFR-mutant lung cancer and KIT-mutant GIST where acquired resistance to the front-line kinase inhibitor is also associated with mutations in the target kinase.104,105
The clinical development of kinase inhibitor combinations to prevent acquired resistance is straightforward. Since the frontline drug is already approved, success would be determined by an improvement in response duration using the combination. The situation is more complex when two experimental compounds (e.g., a PI3K pathway inhibitor and a MEK inhibitor) are combined, neither of which shows significant single agent activity. Current regulatory guidelines require a four arm study that compares each single agent to the combination and to a control group in order to obtain approval of the combination. This design can discourage drug developers as well as patients from moving forward because it requires a large sample size, and it requires that 75% of the patients be assigned to treatment arms that are expected to be inactive. In response to growing preclinical evidence supporting the unique activity of certain combinations, these guidelines are now under revision. A more challenging issue may be optimization of the dose and schedule needed to safely combine two investigational drugs. Much like the development of combination chemotherapy several decades ago, it may be important to select compounds with nonoverlapping toxicities to allow sufficient doses of each drug to be achieved. Investigators may also need to adopt higher toxicity thresholds.
SPECULATIONS ON THE FUTURE ROLE OF KINASE INHIBITORS IN CANCER MEDICINE
The role of genomics in predicting response to kinase inhibitor therapy is now irrefutable. As the number of kinase driver mutations continues to grow, the field is likely to move away from the current strategy of a “companion diagnostic” for each drug. Rather, comprehensive mutational profiling platforms that query each tumor for hundreds of potential cancer mutations are more likely to emerge as the diagnostic platform. Only a small number of mutations are “actionable” in 2010 (meaning the presence of a mutation defines a treatment decision), but this number will undoubtedly grow. In addition, the presence or absence of concurrent mutations will guide decisions about combination therapies.
More effort must be devoted to manipulating the dose and schedule of kinase inhibitor therapy to maximize efficacy and minimize toxicity. To date all kinase inhibitors have been developed based on the assumption that 24/7 coverage of the target is required for efficacy. Consequently, most compounds are optimized to have a long serum half-life (12 to 24 hours). Phase 2 doses are then selected based on the maximum tolerated dose determined with daily administration. But a recent clinical trial of the ABL inhibitor dasatinib in CML indicates that equivalent antitumor activity can be achieved with intermittent therapy.106 By giving larger doses intermittently, higher peak drug concentrations were achieved that resulted in equivalent and possibly superior efficacy.107 Similar results were observed in laboratory studies of EGFR inhibitors in EGFR-mutant lung cancer. Clinically robust, quantitative assays of target inhibition are needed to hasten progress in this area.
Although the focus of this chapter is kinase inhibitors, the themes developed here should apply broadly to inhibitors of other cancer targets. Early clinical results with inhibitors of the G-protein coupled receptor smoothened (SMO) in patients with metastatic basal cell carcinoma or medulloblastoma establish that the driver mutation hypothesis extends beyond kinase inhibitors. SMO is a component in the hedgehog pathway that is constitutively activated in subsets of patients with basal cell carcinoma and medulloblastoma due to mutations in the hedgehog ligand binding receptor Patched-1. Treatment with the SMO inhibitor GDC-0449 led to impressive responses in basal cell carcinoma and medulloblastoma patients whose tumors had Patched-1 mutations.108,109 Other novel cancer targets are emerging from cancer genome sequencing projects. Somatic mutations in the Krebs cycle enzyme isocitrate dehydrogenase (IDH1/2) were found in subsets of patients with glioblastoma and acute myeloid leukemia.110–112 Mutations in enzymes involved in chromatin remodeling were recently found in kidney cancer.113 Drug discovery programs are already under way to find inhibitors of several of these mutant enzymes. Kinase inhibitors are just the first wave of molecularly targeted drugs ushered in through understanding of the molecular underpinnings of cancer cells. There is much more to follow.
Selected References
The full list of references for this chapter appears in the online version.
1. Sawyers CL. Shifting paradigms: the seeds of oncogene addiction. Nat Med 2009;15(10):1158.
2. Weinstein IB. Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science 2002;297(5578):63.
3. Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344(14):1031.
4. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279(5350):577.
6. Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347(7):472.
7. Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 2006;355 (26):2733.
8. Kris MG, Natale RB, Herbst RS, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003;290(16):2149.
10. Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304(5676):1497.
11. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N Engl J Med 2004;350(21):2129.
12. Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004;101(36):13306.
14. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005;353(2):123.
19. Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med 2005;353(2):133.
20. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361(10):947.
23. Cools J, DeAngelo DJ, Gotlib J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003;348(13):1201.
27. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417(6892):949.
28. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005;365(9464):1054.
29. James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434(7037):1144.
30. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005;7(4):387.
31. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004;304(5670):554.
37. Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006;439(7074):358.
45. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363(9):809.
46. Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010;467:596.
51. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448(7153):561.
61. Sawyers CL. Finding the next Gleevec: FLT3 targeted kinase inhibitor therapy for acute myeloid leukemia. Cancer Cell 2002;1(5):413.
67. Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009;114(14):2984.
70. Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349(5):427.
71. Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356(2):125.
72. Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356(2):115.
76. Hudes G, Carducci M, Tomczak P, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 2007;356(22):2271.
77. Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372(9637):449.
81. Thomas GV, Tran C, Mellinghoff IK, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med 2006;12(1):122.
82. Hess G, Herbrecht R, Romaguera J, et al. Phase III study to evaluate temsirolimus compared with investigator,s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009;27(23):3822.
86. Neshat MS, Mellinghoff IK, Tran C, et al. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A 2001;98(18):10314.
87. Majumder PK, Febbo PG, Bikoff R, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med 2004;10(6):594.
88. Podsypanina K, Lee RT, Politis C, et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/– mice. Proc Natl Acad Sci U S A 2001;98(18):10320.
89. O,Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006;66 (3):1500.
93. She QB, Halilovic E, Ye Q, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell 2010;18(1):39.
94. Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008;14(12):1351.
95. Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001;293(5531):876.
96. Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002;2(2):117.
97. Shah NP, Tran C, Lee FY, et al. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004;305(5682):399.
98. Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 2006; 354(24):2531.
99. Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2010;362(24):2260.
100. Sawyers CL. Even better kinase inhibitors for chronic myeloid leukemia. N Engl J Med 2010;362(24):2314.
101. Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 2010;362(24):2251.
104. Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005;2(3):e73.
105. Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 2005;11(11):4182.
106. Shah NP, et al. Intermittent target inhibition with dasatinib 100 mg once daily preserves efficacy and improves tolerability in imatinib-resistant and -intolerant chronic-phase chronic myeloid leukemia. J Clin Oncol 2008;26(19):3204.
107. Shah NP, Kasap C, Weier C, et al. Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer Cell 2008;14(6):485.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


