FIGURE 22.1 Genomic aberrations in prostate cancer. Regions of amplification (red) or deletion (blue) with false discovery rate (FDR) 10% or less are plotted, with chromosomes indicated at the center and centromeres in red. Genes with somatic nonsynonymous mutations are listed on top (black). Additional genes of interest targeted by copy-number alterations alone are also indicated (gray) (From ref. 21 with permission).
Although many individual genes are rarely affected in prostate cancer, several key oncogenic and tumor suppressor pathways are commonly involved when the individual genes in each pathway are considered collectively. Four pathways—the phosphatidylinositol 3-phosphate kinase (PI3K)/AKT pathway, the RAS/MAP kinase pathway, the retinoblastoma pathway, and the androgen receptor (AR) pathway—are altered in one-third of primary cancers and almost all of metastatic lesions. This suggests that in prostate cancer, different individual genes in the pathway may be targeted to activate or suppress a common pathway lesion (Fig. 22.2). The sections below focus on those pathways where prostate cancer is most validated.
Androgen Receptor Pathway
It has been 70 years since Higgins and Hodges made the seminal observation that prostate cancers regress after surgical orchiectomy or suppression of androgen production by administration of exogenous estrogens.22 These observations proved that prostate cancer is an androgen-dependent malignancy, and androgen deprivation therapy became the first targeted therapy in oncology—leading to a Nobel Prize for Charles Huggins in 1966.

FIGURE 22.2 Alterations in the retinoblastoma (A), phosphatidylinositol 3-kinase (PI3K) (B), RAS/MAP kinase (C), and androgen receptor (AR) (D) pathways in prostate cancer. Alteration frequencies are shown for individual genes and for the entire pathway in primary and metastatic tumors. Alterations are defined as those having significant up- or down-regulation compared with normal prostate samples, or by somatic mutations, and are interpreted as activation (red) or inactivation (blue) of protein function. (From ref. 21 with permission.)
Androgens exert their cellular actions through the AR, a 110 kb steroid receptor transcription factor, located on Xq12.23,24 Upon binding androgen, AR mediates transcription of a number of genes involved in survival and differentiation of prostate epithelial cells. AR activity is required for development of both normal and malignant prostate tissue as men with germline AR inactivating mutations or men orchiectomized at a young age do not develop a prostate gland. AR plays a critical role in early pathogenesis as well as progression to advanced disease, and drugs that inhibit AR function remain the primary treatment for advanced prostate cancer.
Early Pathogenesis
Several lines of evidence suggest that enhanced AR signaling may play a causative role in prostate cancer initiation. Polymorphisms of three genes in the AR pathway, AR itself, CYP17, and SRD5A2, are low penetrant risk factors for prostate cancer development in some but not all studies.6 Overexpression of AR in both mouse and human primary prostate cells confers tumor forming ability when injected orthotopically into mice.25,26 In rat models of carcinogen-induced prostate cancer, implantation of testosterone pellets results in an increased number of tumors.27 Transgenic mice that overexpress AR in the prostate develop prostatic intraepithelial neoplasia (PIN).28
Pathway analysis from a recent large-scale prostate cancer genomic project found AR pathway alterations in 60% of primary tumors. The most commonly altered AR pathway member was NCOA2 (also called SRC2), an AR coactivator that potentiates AR transcriptional output. Copy gains or somatic mutations of the NCOA2 gene were found in approximately 20% of primary tumors. In addition, several other AR coactivators showed increased expression, while AR corepressors were down-regulated (Fig. 22.2D).
Cancer Progression and Castration-Resistant Growth
Because of the dependence of the prostate lineage on AR function, chemical castration through suppression of testicular function has become the mainstay in systemic treatment of prostate cancer. In normal prostate tissue, castration results in atrophy and cessation of PSA production indefinitely until restoration of testosterone levels. However, prostate cancers progress to the lethal castration-resistant prostate cancer (CRPC) after a variable duration of response. Cancer progression is usually accompanied by restoration of PSA secretion, indicating reactivation of AR activity. Thus, instead of bypassing the requirement for AR function, most castrate-resistant tumors have reactivation of the AR pathway, likely through multiple mechanisms.23
Alterations of AR itself represent the best characterized mechanism of castration resistance. Mutations of AR are detected in up to 10% of CRPCs and result in activation by other hormone ligands such as corticosteroids or, paradoxically, by antiandrogens.29 Overexpression of AR is seen in the majority of CRPCs, and the AR gene is amplified in approximately 30% of cases.30–32 AR overexpression is sufficient to activate AR in the milieu of castrate levels of androgens and to convert the antiandrogen, bicalutamide, into a weak agonist.33 Alternative splicing of AR mRNA can lead to a truncated protein containing the N-terminal transcriptional activation and DNA binding domains but missing the ligand binding domain. Some of these variants have constitutive transcriptional activity in the absence of androgens and can be detected in CRPC samples. Overexpression of at least two variants can confer castration resistance in preclinical models.34–36
Alterations in AR pathway genes are found in 100% of metastases and may enhance AR activity in the presence of castrate androgen levels. NCOA2, for example, increases the magnitude of AR signaling across a broad range of androgen concentrations. Several other androgen receptor coactivators, including NCOA1, TNK2, and EP300, are up-regulated in metastatic disease, while AR corepressors, including NRIP1, NCOR1, and NCOR2, are down-regulated (Fig. 22.2D).
Intratumoral androgens are not completely eliminated by castration and may be increased in CRPC.37 Direct measurements of intratumoral androgens by mass spectrometry showed that castration-resistant metastatic tumors in men treated with gonadotropin-releasing hormone (GnRH) agonists such as leuprolide have higher levels of testosterone than primary tumors in untreated men.38 There is some evidence that CRPC may synthesize androgens to activate AR in an autocrine loop. Two expression profiling studies that compared metastatic CRPC with primary tumors have shown that enzymes involved in androgen synthesis are up-regulated in CRPC. Holzbeierlein et al.32 found overexpression of enzymes involved in synthesis of cholesterol, the common steroid precursor, from acetyl-CoA, and Stanbrough et al.39 found overexpression of enzymes involved in synthesis of testosterone and the more potent androgen dihydrotestosterone (DHT) from cholesterol. A third study of patient samples at all stages of disease found an abundance of the enzymes AKR1C3 and SRD5A1 necessary for conversion of androstenedione to DHT. However, the samples lacked high expression of enzymes necessary for de novo steroidogenesis.40 These data suggest that autocrine androgen synthesis may allow tumors to grow despite low serum androgen levels, but that this process may in part be dependent on adrenal precursors (Fig. 22.3).
Multiple kinase signaling pathways have been implicated in CRPC—most notably the HER2 and AKT pathways—with evidence that they exert their effects, in part, through modulation of AR activity. The HER2 receptor tyrosine kinase is progressively overexpressed in more advanced CRPC, though it is seldom amplified as seen in breast cancer. In experimental systems, forced overexpression of HER2 results in castration resistance, while pharmacologic inhibition or protein knockdown results in growth suppression.41 Loss of PTEN and activation of the AKT pathway, commonly seen in CRPC (Fig. 22.2B), can contribute to castration resistance. Primary tumors with loss of PTEN have lower response rates to neoadjuvant bicalutamide.42 Prostate cancer in mice with genetically engineered loss of PTEN rapidly develop castration resistance, and PTEN knockdown in murine hormone-sensitive prostate cancer cells confers castration-resistant growth.43,44
Androgen Receptor Targeting
Androgen deprivation therapy (ADT), most commonly delivered by administration of GnRH such as leuprolide, remains the cornerstone of prostate cancer systemic therapy. With the discovery that CRPC is associated with reactivation of AR and remains AR dependent, there have been many recent efforts to develop alternative AR targeting strategies (Fig. 22.3).
Antiandrogens
Antiandrogens compete with endogenous androgens for the ligand binding pocket of AR. Similar to androgens, antiandrogens promote translocation of AR from the cytoplasm into the nucleus and DNA binding but induce conformational changes that prevent optimal transcriptional activity. In the United States, there are three nonsteroidal antiandrogens currently in use—flutamide, bicalutamide, and nilutamide. Although generally active upon treatment initiation, each compound has been associated with the antiandrogen withdrawal response, whereby treated patients with progressive disease derive clinical benefit when the antiandrogen is stopped,45 indicating that they have potential to act as AR agonists despite their intended role as antagonists. Indeed, overexpression of AR can convert bicalutamide from an antagonist into an agonist.33
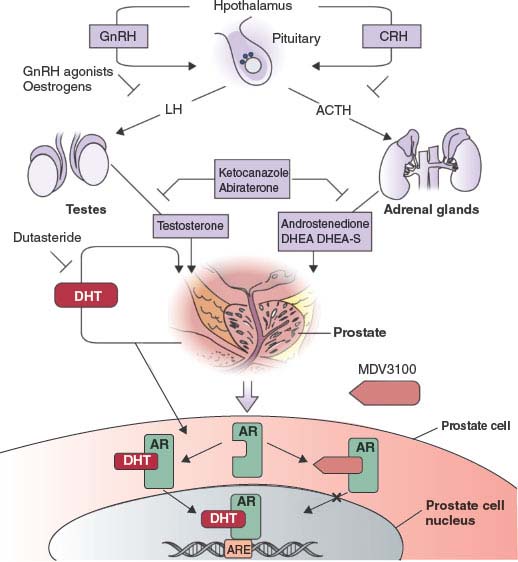
FIGURE 22.3 The androgen-signaling axis and its inhibitors. Testicular androgen synthesis is regulated by the gonadotropin-releasing hormone–luteinizing hormone (GnRH–LH) axis, whereas adrenal androgen synthesis is regulated by the corticotrophin-releasing hormone (CRH)-adrenocorticotropic hormone (ACTH) axis. GnRH agonists and corticosteroids inhibit stimulation of the testes and adrenals, respectively. Abiraterone inhibits CYP17, a critical enzyme in androgen synthesis. MDV3100 competitively inhibits the binding of androgens to androgen receptors. DHEA, dehydroepiandrosterone; DHEA-S, dehydroepiandrosterone sulphate; DHT, dihydrotestosterone; AR, androgen receptor; ARE, androgen-response element. (From ref. 98 with permission.)
Novel antiandrogens that have no agonistic properties would likely be clinically superior, and several efforts to identify such compounds are ongoing. Currently, the most promising compound, MDV3100, binds to AR with high affinity and prevents AR binding to DNA. MDV3100 is highly active in preclinical models of castration resistance where bicalutamide functions as an agonist.46 In a phase 1/2 trial of MDV3100 in 140 CRPC patients previously treated with bicalutamide, the clinical response rate was greater than 50% as measured by a sustained, greater than 50% decline in serum PSA and radiographic evidence of partial response or stable disease. Among those who where chemotherapy naive, PSA response rate was 62% and time to progression was 41 weeks—both compare favorably to docetaxel chemotherapy. Among the more advanced patients refractory to docetaxel, PSA response rate was 51% and time to progression was 21 weeks.47 A phase 3 trial of MDV3100 in men with chemotherapy-refractory CRPC is ongoing.
Androgen Lowering Therapies
The most commonly used form of ADT, leuprolide, inhibits testicular androgen synthesis but does not impact other sources of androgen sources such as the adrenal glands and the purported autocrine androgen synthesis by tumor cells. Second-line ADT agents such as aminoglutethimide and ketoconazole inhibit adrenal androgen synthesis and have modest activity in CRPC, but their clinical utility is limited by side effects due to off target activities that preclude dose escalation.
CYP17 is a key P450 enzyme in the androgen biosynthesis pathway that functions in the testes and adrenal glands to catalyze the conversion of pregnenolone and progesterone into the weak androgens dehydroepiandrosterone (DHEA) and androstenedione, respectively. These weak androgens are further converted into testosterone and DHT, which may occur in peripheral tissues and in prostate tumor cells. Therefore, specific inhibition of CYP17 should decrease androgen synthesis without affecting the production of other essential steroids. The experimental drug abiraterone acetate is a pregnenolone derivative that is a selective, high affinity (half maximal inhibitory concentration [IC50] = 2 nM), irreversible inhibitor of CYP17. In phase 2 studies, two-thirds of chemotherapy-naive and half of postdocetaxel patients had PSA responses.48–50 One potentially undesirable consequence of abiraterone treatment is increased production of adrenal progestins (generated by enzymes upstream of the CPY17 block). These progestins may have agonist activity in CRPC patients whose tumors have a highly sensitized AR pathway. Indeed, addition of dexamethasone to suppress this adrenal activity (and to rescue from potential adrenal insufficiency) reversed abiraterone resistance in one-third of patients (Fig. 22.3).48 Phase 3 trials of abiraterone in men with CRPC are ongoing.
Indirect Approaches
AR activity requires the cooperation of numerous other proteins, some of which can be pharmacologically targeted. AR is stabilized by binding to the molecular chaperone, heat shock protein 90 (HSP90). Indeed, HSP90 inhibitors emerged as hits in a broad screen for chemical compounds that inhibit AR signaling. The clinical HSP90 inhibitor 17-AAG destabilizes AR and causes regression of prostate cancer in preclinical models.51,52 To activate transcription, AR requires coordinated activity of chromatin remodeling enzymes, including histone deacetylases (HDAC). HDAC inhibitors suppress AR activity and prostate cancer growth in preclinical models.53,54 Despite these promising preclinical results, clinical trials of HSP90 and HDAC inhibitors in CRPC have been disappointing, but this may be due to limitations in dose and schedule of these early generation inhibitors.55
ETS TRANSCRIPTION FACTORS
In 2005, using bioinformatic analysis of prostate cancer gene expression data, Tomlins et al.56 made the seminal discovery that one of several members of the E26 transformation-specific (ETS) transcription factors are translocated in over half of all prostate cancers. This discovery also led to the recognition that recurrent genomic translocations, which had previously been observed only in hematopoietic malignancies and sarcomas, can occur commonly in solid tumors. Four ETS members, ERG, ETV1, ETV4, and ETV5, have been reported to be targeted by translocations in prostate cancer.56–58 The typical translocation does not result in a fusion protein. Instead, the translocation usually results in aberrant expression of the targeted ETS factor in the prostate by juxtaposition to the promoter of highly expressed prostate gene—similar to the translocations of MYC and BCL2 to IgG locus in Burkitt and follicular lymphomas (Fig. 22.4). The most prevalent translocation is between the androgen-regulated TMPRSS2 gene and ERG, occurring in approximately 50% of all prostate cancers. Both genes are located on 21q22, and the fusion frequently occurs due to an interstitial deletion, which can be detected by high-density CGH approaches.21

FIGURE 22.4 Schematic of TMPRSS2-ERG and ETV1 fusions. A: TMPRSS2 and ERG are separated by 3Mb on 21q22. TMPRSS2-ERG fusion is usually formed by interstitial deletion resulting in exon 2 of TMPRSS2 juxtaposed to exon 4 of ERG. Fusion results in slightly truncated protein starting from exon 4. B: ETV1 located on 7p21, can be fused to a number of genomic loci, some of which are shown. Many fusions result in a truncated protein starting in exon 6 but full length protein is also observed. Arrow indicates ATG translation initiation sites.
ETS Fusions as Prognostic and Predictive Factors
The discovery of the ETS translocations immediately raised questions about their prognostic value, which are still under debate. Just prior to the discovery of the translocation, one study noted ERG mRNA overexpression in a large subset of prostate cancers (almost certainly a consequence of the TMPRSS2-ERG translocation) reported a favorable prognosis.59 Yet, presence of the TMPRSS2-ERG fusion (detected by fluorescence in situ hybridization [FISH]) strongly predicted for increased disease-specific mortality in a watchful waiting cohort from Sweden.60 Larger studies (of U.S. populations) have found no prognostic role of ERG translocation.61,62 It has been more difficult to assess the effect of the other, non-ERG ETS factors on prognosis due to the much smaller number of affected patients, although several reports consistently report that ETV1 overexpression is associated with poor prognostic features.61,63,64
Given that the most common ETS fusions are expressed under the control of the androgen-regulated TMPRSS2 promoter, their presence may be predictive of response to androgen deprivation therapy. In one small study, ERG rearrangement was associated with improved response to abiraterone acetate, but larger confirmatory studies are needed.65
ETS Fusions Occur Early in the Pathogenic Process
Analysis of primary and metastatic human tumors indicates that the TMPRSS2-ERG fusion (and most likely the rarer ETV fusions) are early events in prostate oncogenesis. Interestingly, independent prostate cancer loci in the prostatectomy sample from a single patient may harbor different types of ETS fusions, raising the possibility of multiple, concurrent primaries. However, studies of metastatic lesions uniformly contain the same type of ETS fusion, suggesting that the fusion is a premetastatic event and that not all fusion-positive tumors progress.66–68 This pattern contrasts with genomic PTEN loss or AR amplification where the prevalence increases in castration-resistant metastatic lesions, suggesting these may represent later events. The role of ETS fusions in prostate cancer initiation is more controversial, particularly since mice engineered to express ETS fusions in the prostate do not develop cancers. Using FISH, early studies suggests that ETS fusions are rare in PIN. However, immunohistochemistry studies with a highly specific ERG antibody reveal that PIN lesions associated with ERG-positive carcinoma are generally positive for ERG.69
Oncogenic Role of ETS Factors
ETS factors are atypical oncogenes in that they can be highly expressed in some normal tissues (e.g., ERG in endothelial cells and ETV1 in neurons). Furthermore, forced ETS overexpression in cells in culture is usually not transforming. Like many transcription factors, aberrant expression under specific contexts is likely required for oncogenesis. In the prostate, activation of the PTEN/AKT pathway provides one context for ETS-mediated tumorigenesis. In mouse models, overexpression of ERG or ETV1 alone in the prostate has only modest effects.56,70,71 Yet, in the context of PTEN loss or AKT activation, ERG overexpression is oncogenic. Indeed, in human prostate cancers, there is a strong correlation between ERG overexpression and loss of PTEN.72–74
The transcriptional program governed by aberrantly overexpressed ETS in prostate cancer is currently not well understood. To address this question, the genomic binding sites of ERG have recently been mapped in prostate cancer. Surprisingly, there is an extraordinarily high overlap between ERG and AR binding sites—suggesting that ERG can modulate AR activity.75 Since AR can promote both cellular growth and differentiation, one hypothesis is that ERG expression modulates AR function away from differentiation and toward proliferation.
The PTEN/AKT Pathway
The PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor gene, originally identified through studies in breast cancer and glioblastoma, is lost in a significant fraction of prostate cancers through deletion, mutation, or epigenetic silencing.76 Studies using high-density SNP arrays show that PTEN is the only gene in the overlapping chromosomal regions lost in a large set of prostate cancers displaying deletion of 10q23.77 Examination of nine prostate cancer xenografts with neither PTEN loss of heterozygosity (LOH) nor mutations showed that five had epigenetic silencing.78 Three independently generated mouse models with prostate-specific homozygous deletions of Pten all developed prostate cancer with progression from PIN to adenocarcinoma to metastasis similar to human disease.79–81 These data unambiguously implicate PTEN as a critical lesion in prostate cancer pathogenesis. Whether PTEN loss occurs early or late in prostate tumorigenesis is not clear. Complete genomic loss of PTEN is seen in greater than 50% of metastatic lesions but in only approximately 20% of localized lesions. Within the same patient, PTEN loss can be observed in metastatic but not primary lesions,68 suggesting that genomic PTEN loss is involved in disease progression but not initiation. However, heterozygous PTEN loss or decreased PTEN protein expression by immunohistochemistry has been reported in approximately 25% of PIN and greater than 50% of primary cancers, suggesting that PTEN may be a haploinsufficient tumor suppressor. Partial loss could be important for early tumorigenesis, with progression to complete loss at metastasis.82 Mouse models have confirmed a dosage effect of Pten in prostate cancer progression.
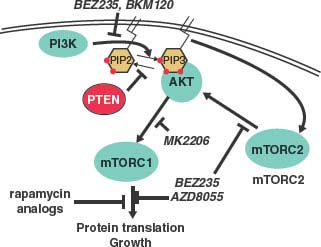
FIGURE 22.5 Schematic of the phosphatidylinositol 3-kinase (PK3K) pathway and inhibitors. PI3K phosphorylates PIP2 into PIP3, while the PTEN phosphatase catalyzes the opposite reaction. PIP3 activates AKT by recruitment to the membrane, activation of mammalian target of rapamycin complex 2 (mTORC2) and pyruvate dehydrogenase kinase (PDK) kinases. AKT activates many growth pathways include mTORC1. BEZ235 is a dual-specificity kinase inhibitor that inhibits both PI3K and mTOR, the common kinase component of mTROC1 and mTORC2 complexes. BKM120 and AZD8055 are kinase inhibitors that inhibit PI3K and mTOR, respectively. Rapamycin analogues (e.g., sirolimus, temsirolimus, everolimus) specifically inhibit assembly of the mTORC1 complex. MK2206 is a new generation allosteric inhibitor of AKT.
PTEN is the major negative regulator of the PI3K/AKT pathway (Fig. 22.5). PI3K, which can be activated by many growth factors, phosphorylates PIP2 into PIP3 at the plasma membrane and PIP3 recruits pleckstrin homology (PH) domain–containing proteins, including AKT. As a lipid phosphatase, PTEN converts PIP3 back to PIP2 thereby limiting normal activity of the pathway. PI3K also activates mammalian target of rapamycin (mTOR) C2 complex (which contains the kinase mTOR complexed with distinct regulatory factors), which directly phosphorylates and activates AKT. Activated AKT phosphorylates a number of crucial substrates implicated in cell growth and survival, including downstream activation of the other mTOR complex, mTORC1 (Fig. 22.5).
The PTEN→AKT→mTOR pathway has garnered special attention due to the large number of mutations in the genes in this pathway, including PTEN, PI3K, AKT, as well as upstream kinases. Mouse models of prostate cancer suggest that both AKT and mTORC2 are essential mediators of PTEN-loss–mediated tumorigenesis—prostate-specific deletions of either gene abolish the tumor phenotype of Pten loss prostate.83,84 This pathway is also amenable to pharmaceutical inhibition using approved drugs such as rapamycin, and its analogues, which inhibit mTORC1; investigational kinase inhibitors targeting PI3K, mTORC1 and mTORC2, and AKT are in clinical development (Fig. 22.5). Preclinical data provide strong rationale for their trials in prostate cancer. In addition to AKT, PTEN loss likely has other crucial targets, evidenced by the observation that overexpression of active AKT in mice causes PIN that does not progress into cancer while Pten(–/–) mice develop invasive cancer. One candidate pathway involves CDC42 and JNK.85
Chromosome 8p Loss and 8q Gain
NKX3.1
Heterozygous loss of 8p21 is the most frequent chromosomal aberration in prostate cancer, affecting 60% of PIN and 85% of prostate cancers.86 The most studied candidate tumor suppressor gene mapping to this region is the AR regulated, prostate specific homeobox transcription factor NKX3.1. However, unlike classic tumor suppressor genes, the remaining NKX3.1 allele is not mutated or deleted, leading to the hypothesis that NKX3.1 haploinsufficiency is sufficient for oncogenesis. Prostate development in mice with targeted deletion of Nkx3.1 proceeds normally, albeit with subtle defects in prostate morphogenesis.86 Mechanistic studies suggest NKX3.1 facilitates terminal differentiation. In castrated mice, introduction of testosterone results in an early intense proliferative response to reconstitute the gland followed by differentiation and quiescence. In Nkx3.1(+/–) and Nkx3.1(–/–) mice, there is a delay in differentiation, resulting in hypertrophy.87,88 When crossed into Pten(+/–) mice, loss of Nkx3.1 is reported to accelerate prostate neoplasia. Furthermore, prostate tumors that develop in two genetically engineered mouse models caused by distinct initiating lesions (transgenic expression of Myc and conditional Pten deletion) both show reduced Nkx3.1 expression.79,89 In the case of Pten loss, the decline in Nkx3.1 expression is most likely explained by crosstalk between Pten and Nkx3.1 rather than selection for Nkx3.1 loss as a progression event.90
Other evidence argues against NKX3.1 as the 8p tumor suppressor. Despite monoallelic loss, mRNA expression is still quite high in prostate tumors. In fact, tumors that harbor single copy loss of NKX3.1 do not have lower levels of NKX3.1 transcript. Furthermore, the minimal common deleted region on 8p does not always span NKX3.1. Thus, there may be other tumors suppressors in 8p21 whose loss mediate prostate tumorigenesis.
MYC
Gain of 8q is the second most common chromosomal abnormality in prostate cancer with peaks of amplification located at both MYC and NCOA2. Metastatic lesions often have further amplicons at the MYC locus on 8q24. MYC is a transcription factor that heterodimerizes with Max to regulate the expression of a large number of genes (up to 15% of the genome) and serves as an integrator of numerous growth signals. Physiologically, MYC expression correlates with proliferation, and levels are tightly controlled at multiple levels—by transcription, mRNA stability, and protein stability. There is strong experimental evidence implicating MYC dysregulation in prostate cancer. MYC overexpression in primary prostate epithelial cells leads to transformation,25 and transgenic mice expressing MYC in the prostate develop PIN followed by adenocarcinoma,89 which histologically represents the closest mimic of human prostate cancer among current mouse models.91
Epigenetic Changes
In addition to genetic alternations, a number of epigenetic changes have been implicated in prostate cancer progression, such as altered DNA methylation and histone modifications, that lead to changes in gene expression.92 One early change in prostate cancer is silencing of the π-class glutathione S-transferase (GSTP) gene through DNA methylation at CpG islands in transcriptional regulatory regions. GSTP is frequently overexpressed in many cancers, perhaps due to oncogenic and chemotherapy stress. Unexpectedly, GSTπ expression is silenced in the vast majority of prostate cancers and PIN lesions through methylation of the GSTπ promoter.93
DNA is packaged around the core histone proteins H2A, H2B, H3, and H4 to form chromatin. The amino termini of histones, particularly H3 and H4, are subject to posttranslational modification through lysine acetylation or methylation and arginine methylation, all of which influence gene expression. Altered histone modifications, such as H3K4 methylation and H4R3 methylation, have been reported to correlate with reduced risk of recurrence in surgically resected prostate cancer.94 Changes in expression of the proteins responsible for these histone modifications have been implicated in prostate cancer. For example, EZH2 (enhancer of Zeste homolog 2) is a polycomb group protein overexpressed in high-grade localized as well as castrate-resistant metastatic prostate cancer.95 EZH2 is part of a larger suppressive complex that recruits DNA methyltransferases to CpG islands. EZH2 methylates lysine 27 of histone H3 and forms a bridge between suppressive histone and DNA methylation.96 High levels of EZH2 may be oncogenic through suppression of gene expression due to H3K27 methylation of critical target genes such as the RasGAP family member DAB2IP.97
Selected References
The full list of references for this chapter appears in the online version.
6. Nelson WG, De Marzo AM, Isaacs WB. Prostate cancer. N Engl J Med 2003;349:366.
7. De Marzo AM, Platz EA, Sutcliffe S, et al. Inflammation in prostate carcinogenesis. Nat Rev Cancer 2007;7:256.
8. Schroder FH, Hugosson J, Roobol MJ, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med 2009;360:1320.
9. Andriole GL, Crawford ED, Grubb RL 3rd, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med 2009;360:1310.
11. Amundadottir LT, Sulem P, Gudmundsson J, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet 2006;38:652.
12. Gudmundsson J, Sulem P, Manolescu A, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet 2007; 39:631.
13. Haiman CA, Patterson N, Freedman ML, et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet 2007;39:638.
14. Yeager M, Orr N, Hayes RB, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 2007;39:645.
15. Al Olama AA, Kote-Jarai Z, Giles GG, et al. Multiple loci on 8q24 associated with prostate cancer susceptibility. Nat Genet 2009;41:1058.
16. Ahmadiyeh N, Pomerantz MM, Grisanzio C, et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A 2010;107:9742.
17. Eeles RA, Kote-Jarai Z, Giles GG, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet 2008;40:316.
18. Thomas G, Jacobs KB, Yeager M, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet 2008;40:310.
19. Zheng SL, Sun J, Wiklund F, et al. Cumulative association of five genetic variants with prostate cancer. N Engl J Med 2008;358:910.
21. Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18:11.
22. Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res 1941;1:293.
24. Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 2008;8:440.
25. Berger R, Febbo PG, Majumder PK, et al. Androgen-induced differentiation and tumorigenicity of human prostate epithelial cells. Cancer Res 2004;64:8867.
30. Visakorpi T, Hyytinen E, Koivisto P, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet 1995;9:401.
32. Holzbeierlein J, Lal P, LaTulippe E, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol 2004;164:217.
33. Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med 2004;10:33.
36. Sun S, Sprenger CC, Vessella RL, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest 2010;120:2715.
38. Montgomery RB, Mostaghel EA, Vessella R, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 2008;68:4447.
39. Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res 2006;66:2815.
41. Mellinghoff IK, Vivanco I, Kwon A, et al. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell 2004;6:517.
45. Kelly WK, Scher HI. Prostate specific antigen decline after antiandrogen withdrawal: the flutamide withdrawal syndrome. J Urol 1993;149:607.
46. Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324:787.
47. Scher HI, Beer TM, Higano CS, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet 2010;375:1437.
49. Danila DC, Morris MJ, de Bono JS, et al. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol 2010;28:1496.
52. Hieronymus H, Lamb J, Ross KN, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 2006;10:321.
54. Welsbie DS, Xu J, Chen Y, et al. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res 2009;69:958.
56. Tomlins SA, Laxman B, Dhanasekaran SM, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007; 448:595.
58. Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005;310:644.
60. Demichelis F, Fall K, Perner S, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 2007;26:4596.
62. Gopalan A, Leversha MA, Satagopan JM, et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res 2009;69:1400.
72. Carver BS, Tran J, Gopalan A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet 2009;41:619.
73. King JC, Xu J, Wongvipat J, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet 2009;41:524.
74. Zong Y, Xin L, Goldstein AS, et al. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A 2009;106:12465.
75. Yu J, Mani RS, Cao Q, et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 2010;17:443.
78. Whang YE, Wu X, Suzuki H, et al. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A 1998;95:5246.
79. Wang S, Gao J, Lei Q, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003;4:209.
83. Chen ML, Xu PZ, Peng XD, et al. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev 2006;20:1569.
84. Guertin DA, Stevens DM, Saitoh M, et al. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009;15:148.
85. Vivanco I, Palaskas N, Tran C, et al. Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell 2007;11:555.
87. Kim MJ, Bhatia-Gaur R, Banach-Petrosky WA, et al. Nkx3.1 mutant mice recapitulate early stages of prostate carcinogenesis. Cancer Res 2002;62:2999.
88. Magee JA, Abdulkadir SA, Milbrandt J. Haploinsufficiency at the Nkx3.1 locus. A paradigm for stochastic, dosage-sensitive gene regulation during tumor initiation. Cancer Cell 2003;3:273.
89. Ellwood-Yen K, Graeber TG, Wongvipat J, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003;4:223.
90. Lei Q, Jiao J, Xin L, et al. NKX3.1 stabilizes p53, inhibits AKT activation, and blocks prostate cancer initiation caused by PTEN loss. Cancer Cell 2006;9:367.
94. Seligson DB, Horvath S, Shi T, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005;435:1262.
95. Varambally S, Dhanasekaran SM, Zhou M, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002;419:624.
98. Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol 2009;10:981.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


