FIGURE 11.1 Models of tumor heterogeneity. Tumors are composed of phenotypically and functionally heterogeneous cells. There are two theories as to how this heterogeneity arises. According to the stochastic model, tumor cells are biologically equivalent but their behavior is influenced by intrinsic and extrinsic factors and is therefore both variable and unpredictable. Thus, tumor-initiating activity cannot be enriched by sorting cells based on intrinsic characteristics. In contrast, the cancer stem cell model postulates the existence of biologically distinct classes of cells with differing functional abilities and behavior. Only a subset of cells has the ability to initiate tumor growth; these cancer stem cells possess self-renewal and give rise to nontumorigenic progeny that make up the bulk of the tumor. This model predicts that tumor-initiating cells can be identified and purified from the bulk non-tumorigenic population based on intrinsic characteristics. (Figure originally published in Dick JE. Stem cell concepts renew cancer research. Blood 2008;112:4793.) © The American Society of Hematology.
CANCER STEM CELLS IN SOLID TUMORS
Breast Cancer Stem Cells
Investigations of mechanisms underlying solid tumor heterogeneity were first undertaken in human breast cancer. Al Hajj et al.15 made single-cell suspensions of breast cancer specimens obtained from primary or metastatic sites in patients. Upon injection into the mammary fat pad of immune-deficient NOD/SCID mice, all samples studied were able to generate tumors. Thus, the NOD/SCID model provides a functional assay of the in vivo tumor-initiating ability of human breast cancer cells. Breast cancer cells are heterogeneous with respect to expression of a variety of surface markers, including the adhesion molecules CD24 and CD44. To test whether it would be possible to identify and isolate subpopulations enriched for tumor-initiating activity, breast cancer cells were first separated from normal hematopoietic, endothelial, mesothelial, and fibroblast cells by elimination of cells expressing lineage markers, then subfractionated based on expression of CD24 and CD44. All of the in vivo tumor-initiating activity was found in the CD44+CD24−/lowLineage− cell fraction, with enrichment compared to unfractionated tumor cells as judged by the cell dose required for tumor formation. CD44− and CD44+CD24+Lineage− cells, even though morphologically indistinguishable from tumor-initiating cells, did not generate tumors at injection sites. In some samples, isolation of cells expressing epithelial specific antigen (ESA) allowed further enrichment of tumor-initiating activity within the CD44+CD24−/lowLineage− population, however, ESA expression did not distinguish between tumorigenic and nontumorigenic cells in at least one sample, and therefore may not be a reliable marker for breast cancer-initiating cells. CD44+CD24−/lowLineage− tumorigenic cells could be serially propagated, demonstrating self-renewal, and gave rise not only to more CD44+CD24−/lowLineage− tumorigenic cells, but also to the phenotypically diverse nontumorigenic cells which made up the bulk of the primary tumor, thereby recapitulating the tumor’s complexity and functional heterogeneity. This study was the first to isolate tumor-initiating cells from the bulk nontumorigenic population in a nonhematological malignancy, providing strong evidence that the growth of at least some types of human solid tumors is sustained by biologically distinct CSCs.
Other investigators have since shown that breast CSCs can be isolated from some patient tumors by cellular expression of aldehyde dehydrogenase (ALDH), with partial overlap with the CD44+CD24−Lineage− phenotype.16 As few as 20 ALDH1+CD44+CD24−Lineage− cells from one patient cancer could generate tumors in NOD/SCID mice. Although highly enriched, these CSC-containing fractions are still heterogeneous, and additional CSC markers will be required for further purification. Nevertheless, identification of breast CSCs has moved cancer researchers away from studying bulk tumors and shifted their focus to understanding the biology of this subpopulation of cells. For example, Yu et al.17 have shown that cell fractions enriched for breast CSCs have globally reduced microRNA expression compared to more differentiated cancer cells. In particular, the let-7 family is not expressed and increases with differentiation. Lentiviral expression of let-7 in Lineage−CD44+CD24− cells from patient breast cancers significantly reduced tumor formation in both primary and serially transplanted NOD/SCID mice. Insight into the biology of CSCs will be a crucial first step to develop an effective means to target them therapeutically.
Brain Cancer Stem Cells
Studies in several types of human brain cancers have clearly shown that the tumor cell population is functionally heterogeneous, in that only a fraction of cells have the ability to form tumor neurospheres when plated at low density in culture or generate tumors when transplanted in vivo.18,19 As discussed above, demonstration that this heterogeneity arises from the existence of biologically distinct cell populations, rather than as a result of stochastic processes, requires isolation of tumor-initiating cells from the bulk nontumorigenic population. Singh et al.20 reported that CD133+ cells in different types of human brain tumors possess extensive proliferative, differentiative, and self-renewal capacity in vitro. The development of a xenograft assay that involved injection of single-cell suspensions of human brain tumor samples into the NOD/SCID mouse brain enabled assessment of whether the CD133+ cells were capable of initiating tumor growth in vivo.21 Tumors could be generated by as few as 100 CD133+ cells, while injection of up to 105 CD133− tumor cells did not result in tumor formation. Importantly, small numbers of viable CD133− tumor cells could be found at the injection site many weeks later, ruling out the possibility that CD133− cells did not form tumors simply because they died following transplantation. The tumors generated by CD133+ cells resembled the patient’s original tumor by immunohistochemistry and consisted of a minority CD133+ and a majority CD133− cell population. CD133+ cells isolated from xenograft tumors could generate phenotypically similar tumors in secondary mice. Thus, CD133+ cells from human brain tumors possess the two key properties of CSCs: the ability to self-renew and to recapitulate tumor heterogeneity through differentiation. Interestingly, CD133 (also called prominin-1/AC133) has also been used as a marker to enrich normal human HSCs as well as stem cells in the human central nervous system, suggesting that it may be a marker of both normal and malignant stem cells.
Recently, there have been reports that CD133 may not be a universal marker of CSCs in brain cancer.22–24 In addition, a study of neurosphere lines derived from PTEN-deficient human glioblastomas found that both CD133+ and CD133− cells could generate serially transplantable tumors in xenograft recipients and provided evidence that these tumors comprise a hierarchy of self-renewing CSC populations with variable tumorigenic capacity.25 Unfortunately, these investigators were not able to study sorted cell populations directly isolated from patient tumors. Cell surface phenotype can change significantly during culture, without corresponding changes in stem cell function.26 In addition, there is evidence that CD133 expression can vary as a function of cell cycle.27 Thus, the results of these studies should be interpreted with caution. Nevertheless, it would not be surprising to find that the CSC phenotype is heterogeneous, even among tumors of the same histologic subtype, given the variety of genetic and epigenetic perturbations that can ultimately lead to tumor formation. These observations underscore the importance of characterizing candidate CSC populations through functional assays of their tumor-forming ability rather than relying solely on phenotypic identification.
Cancer Stem Cells in Other Solid Tumors
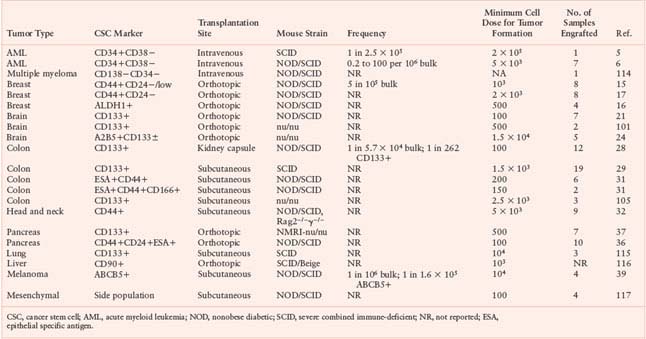
Two groups initially reported isolation of CD133+ tumor-initiating cells from human colon cancers.28,29 Single-cell suspensions of primary or metastatic tumor samples were injected either under the renal capsule of NOD/SCID mice28 or subcutaneously into SCID mice.29 In both studies, only CD133+ and not CD133− cells, which composed the bulk of the cancers, were able to initiate tumor formation in vivo. Tumors could be serially propagated by reisolating CD133+ cells from xenografts and transplanting them into secondary mice. The ability to perform quantitative analysis is an essential feature of any in vivo assay. In one study, the frequency of colon CSCs in the bulk tumor was determined by limiting dilution analysis to be 1 in 60,000 colon cancer cells.28 The frequency of CSCs in the CD133+ cell fraction was 1 in 262, representing a greater than 200-fold enrichment over unfractionated cells. Clearly, however, the majority of CD133+ colon cancer cells are not CSCs. As has been shown in the CD34+ cell fraction of AML,30 there may be a hierarchy of CSCs and progenitors in the CD133+ subpopulation of colon cancer cells. Another study identified colon CSCs in the EpCAMhighCD44+CD166+ cell fraction.31 CD44+ cells generally constituted a minority subset of the CD133+ population, thus CD44 is a marker that could potentially be used to purify the CSC-containing fraction further.
Using methodology similar to that described above, CD44+ CSCs have recently been characterized in squamous cell carcinomas of the head and neck (HNSCC),32 adding to the rapidly growing list of human cancers in which a distinct CSC population has been identified (Fig. 11.2, Table 11.1). Significantly, in moderately to well-differentiated HNSCC in which some tissue architecture is preserved, CD44+ cells were localized to the basal layer and costained with Cytokeratin 5/14, a marker of normal squamous epithelial stem and progenitor cells, but not with the differentiation marker involucrin. Furthermore, CD44+ cells expressed much higher levels of BMI1 than CD44− cells. BMI1 plays an important role in the self-renewal of hematopoietic and neuronal stem cells and has been implicated in tumorigenesis.33–35 These findings demonstrate that biological pathways likely differ between tumorigenic and nontumorigenic populations and underscore the importance of identifying and characterizing the CSCs within tumors, both for gene expression and proteomic analyses as well as therapeutic targeting.
Expression of CD44 on CSCs from both breast cancer and HNSCC suggests that this adhesion molecule may also be a marker of CSCs in other tumors of epithelial origin. Recently, Li et al.36 showed in pancreatic adenocarcinoma that cell fractions expressing CD44, CD24, epithelial specific antigen (ESA), or a combination of these markers were enriched for tumorigenic activity, as assessed by the frequency of tumor formation following subcutaneous or intrapancreatic injection into NOD/SCID mice. The most highly enriched fraction comprised cells that express all three of these markers, with tumor formation in half of mice receiving as few as 100 CD44+CD24+ESA+ cells. However, injection of CD44−, CD24−, or ESA− cells also gave rise to tumors, albeit with lower frequency. Thus, none of the markers used in this study enabled clear separation of cells with tumor-initiating activity from the bulk nontumorigenic population. In contrast to these results, Hermann et al.37 identified CSCs in pancreatic tumors by CD133 expression; as few as 500 freshly isolated CD133+ cells were able to generate tumors after orthotopic injection into immune-deficient mice, whereas as many as 1 × 106 CD133− cells could not.
The list of tumors in which CSC populations have been identified continues to grow (Table 11.1). There is frequent overlap in cell surface phenotype among CSCs from different tumor types, as evidenced in particular by expression of CD44 and CD133 on CSCs from epithelial tumors. Whether these are simply surrogate markers or play a functional role in CSC biology has yet to be determined.
Controversies and Future Directions
There have been a number of criticisms of the CSC model. As discussed above, characterization of CSC populations requires xenotransplantation into immune-deficient recipients. Some investigators have argued that inefficiencies of the xenotransplant system lead to underestimation of the frequency of cells with tumor-initiating ability.38 In fact, when improvements have been made to these assay systems, for example through the use of more immune-deficient recipients such as NOD/SCID Il2rg−/− mice, the frequency of tumor initiating cells is sometimes dramatically increased, as seen recently for melanoma (greater than 4 log difference).39,40 There are a number of issues to be considered here. First, such large increases in CSC frequency with the use of more immune-deficient recipients are not universal—for example, they have not been observed in AML41,42,42a—possibly reflecting variable sensitivity of tumor-initiating cells from different tumor types to residual host immune surveillance. Second, while human cell engraftment in xenotransplant assays is undoubtedly limited by residual elements of the recipient immune system, absence of cross-reactivity of cytokines, and other components of the host microenvironment, the low frequency of tumor-initiating cells seen in some cancers cannot be wholly explained by xenotransplantation barriers. For example, LSC frequency in two murine models of leukemia involving MOZ-TIF expression or Pten deletion was low (1 in 104 to 1 in 6 × 105) despite syngeneic bone marrow transplantation.12,43 Furthermore, in contrast to the low frequency of LSCs generally seen when human AML cells are xenotransplanted, much higher LSC frequencies (on the order of 1%) have been observed in a genetically induced model of human B-cell acute lymphoblastic leukemia (ALL) that involves transplantation of human cord blood stem or progenitor cells expressing the MLL-ENL oncogene into immune-deficient mice.42,44 This frequency was comparable to that reported for a murine model of MLL-AF9–induced AML (1 in 150).45 These findings indicate that CSC frequencies can vary widely in different cancers, regardless of whether they are quantified using xeno- or syngeneic transplant assays, and likely relate at least in part to the specific underlying oncogenic pathways operating within the different tumors. Third, and perhaps most important, the CSC hypothesis does not address the absolute frequency of these cells: it is not required that CSCs be rare. The CSC model, at its most fundamental level, simply proposes that the basis of functional heterogeneity within tumors is the existence of cell populations that can be distinguished from other tumor cells by their ability to initiate malignant growth in vivo.
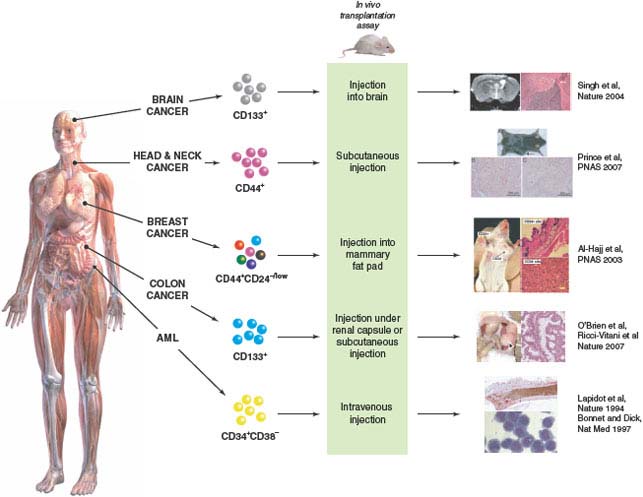
FIGURE 11.2 Identification of cancer stem cells (CSCs) in acute myeloid leukemia (AML) and solid tumors. Subfractions of tumor cells isolated from the bulk tumor population are assayed for their ability to initiate tumor growth in vivo using immune-deficient mice as recipients. CSCs are the only cells capable of initiating tumor growth, giving rise to more CSCs through self-renewal, and also to nontumorigenic differentiated progeny, thus recapitulating the functional heterogeneity of the original tumor. CSCs were first identified in AML and have now also been identified in several types of solid tumors (Table 11.1). (Brain cancer images adapted by permission from Macmillan Publishers Ltd: Nature (21), copyright 2004. Head and neck cancer and breast cancer images copyright 2007 (ref. 32) and 2003 (ref. 15), respectively, National Academy of Sciences, USA. Colon cancer images are from ref. 28.)
TABLE 11.1
PROSPECTIVE ISOLATION OF CANCER STEM CELLS FROM HUMAN LEUKEMIAS AND SOLID TUMORS
It is possible that some human cancers may not follow the CSC model. For example, the high frequency (one in four) and lack of a clear isolatable phenotype of tumor-initiating cells in melanoma suggest the absence of a hierarchical organization.40 Regardless of whether a tumor contains a subpopulation of CSCs responsible for maintaining tumor growth or common tumorigenic cells with little evidence of a CSC hierarchy, all cancer cells with the ability to initiate disease must be identified and targeted therapeutically in order to achieve cure. Since the first published reports of CSCs in breast and brain cancers, there have been numerous studies of CSCs in other solid tumors, with varying degrees of robustness. When weighing evidence presented for or against the CSC hypothesis, one must consider not only the rigor of the xenotransplant assay but also experimental details such as whether cancer cells have been extensively cultured in vitro or passaged in vivo, both of which can lead to changes in function and phenotype, and whether freshly isolated cells from patients’ tumors have been used versus cell lines that may not be representative of clinical disease.
Nevertheless, there is accumulating evidence that the growth of several types of human cancer is initiated and maintained by a subset of phenotypically and functionally distinct CSCs. Although the markers used to date to identify the CSC subset have enabled enrichment of this population compared to unsorted tumor cells, in all cases the enriched cell fractions are still functionally heterogeneous, containing both CSCs and their nontumorigenic progeny. One of the challenges of future research will be to obtain more purified populations of CSCs for use in molecular studies such as gene expression profiling. Novel protocols to purify cells with in vivo tumor-initiating capacity may combine cell surface markers with functional parameters such as Hoechst 33342 dye efflux, which identifies a “side population” of cells with high drug efflux capacity46 or high aldehyde dehydrogenase activity.47 Ultimately, rigorous proof for the existence of biologically distinct CSCs can only be obtained through demonstration that a single cell has the ability to self-renew and to recapitulate the entire tumor hierarchy. This will require either development of in vivo tumor models that support the growth of singly transplanted cells or clonal analysis techniques that enable tracking of the progeny of individually marked tumor cells in vivo, as demonstrated for AML LSCs.30
GENETIC DIVERSITY AND CLONAL EVOLUTION IN CANCER
Most of the initial studies identifying and characterizing CSCs in leukemia and solid tumors did not investigate the underlying genetic changes in cancer cells. Intratumor clonal heterogeneity has been described in many types of cancer48 and may contribute to progression.49 Investigators have proposed that intratumor heterogeneity arises through clonal evolution,48 a long-standing concept in which cancer cells within a tumor acquire various mutations over time, leading to genetic drift and stepwise natural selection for the fittest, most aggressive cells, both of which drive tumor progression.50 According to this model, the parallel growth and contraction of related but divergent subclones result in intratumor heterogeneity, and the genetic makeup of the tumor at any particular time reflects the activity of the dominant subclone(s). Recently, a number of studies have shown that phenotypically defined stem and progenitor populations in human breast cancers are clonally related but not identical.51–53 There is now evidence from two studies in All54,54a that genetically distinct subclones that possess differing growth properties are present even at diagnosis. Examination of the genetic changes acquired by different subclones allowed reconstruction of their ancestry and provided evidence of a complex genetic architecture in ALL, with multiple subclones that are related in a branching rather than linear fashion. Importantly, the genetic diversity found in ALL patient samples was regenerated upon transplantation in immune-deficient mice, suggesting that leukemia-initiating cells are genetically diverse in this disease.
Although the existence of CSCs in most subtypes of human ALL is still under debate, based on these functional studies it may be reasonable to bring together the idea of clonal evolution driving intratumor genetic diversity and the existence of CSCs in a broader model of cancer progression, rather than viewing them as mutually exclusive concepts. Instead of being rigid, linear hierarchies that mirror normal tissue stem cell hierarchies, tumors should be thought of as genetically dynamic, comprising related but divergent subclones driven by CSCs, which are the units of evolutionary selection. On one hand, genetically distinct CSC clones may arise through changes acquired by existing CSCs. For example, the LSCs in an experimentally induced human leukemia can evolve through rearrangement of their immunoglobulin heavy chain genes.44 On the other hand, CSCs may also arise through transformation of non-CSC populations, through acquisition of additional alterations or processes such as epithelial-mesenchymal transition (see below) (Fig. 11.3). Over time the genetic composition of the tumor may change, with different CSC-driven subclones becoming dominant or disappearing, either through natural selection based on cell-intrinsic or microenvironmental factors, or through selection by therapy. Indeed, clonal studies of matched diagnosis and relapse samples from pediatric patients with ALL have shown that relapse clones were often present as minor subpopulations at diagnosis.55 Interestingly, therapy seemed to select for genetic abnormalities related to cell cycle regulation and B-cell development.
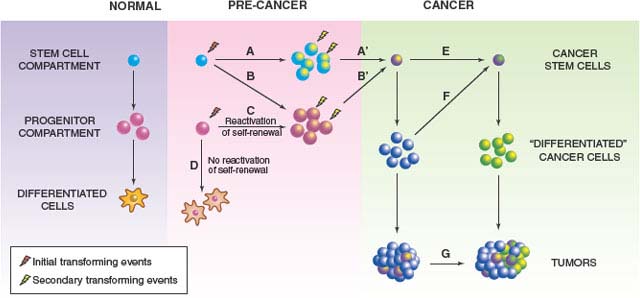
FIGURE 11.3 Models of tumor initiation and progression. Cancer stem cells (CSCs) might arise through neoplastic changes initiated in normal self-renewing stem cells, or downstream progenitors with limited or no self-renewal. Initial events in stem cells could cause (A) expansion of the stem cell pool and/or (B) expansion of downstream progenitors. Secondary events are more likely to occur in these expanded pools of target cells (A’, B’); thus, it is possible that transformation is initiated in a normal stem cell, but the final steps occur in downstream progenitors (BSB’). Reactivation of a self-renewal program is a central feature of oncogenic transformation of progenitors (C), as these short-lived cells will otherwise die or undergo terminal differentiation before enough mutations occur for full neoplastic transformation (D). CSCs themselves might acquire additional genetic or epigenetic changes during tumor progression (E), and non-CSCs can acquire CSC-like properties (F) through processes such as epithelial-mesenchymal transition (EMT), leading to evolution of tumor phenotype (G) and intratumor heterogeneity. Of note, the cell of origin debate centers around the cells and processes illustrated in the pink box, whereas CSC research focuses on characterization of cancer cell populations after they reach the green box. (Figure originally published in Wang JCY. Good cells gone bad: the cellular origins of cancer. Trends Mol Med 2010;16:145.)
If found to be a property of cancer in general, genetic diversity of CSCs could have important implications for the development of anticancer therapies. Regardless of their origin, CSCs are the key cells that drive tumor growth and that must therefore be eliminated in order to achieve cure. If CSCs are genetically, and as a result functionally, diverse, they could represent a moving therapeutic target. Agents that target critical molecular pathways may fail if the selected targets are not present in some CSC subclones or if others have acquired changes that bypass their dependence on the targeted pathways. The development of successful therapeutic regimens will have to take into account the genetic and functional heterogeneity of CSCs.
THE ORIGINS OF CANCER STEM CELLS
A key focus of cancer research is elucidation of the molecular changes that underlie tumor initiation and progression. Tumorigenesis is a multistep process, and CSCs can be regarded as cells that have accumulated enough genetic or epigenetic changes to become fully transformed and that possess a stem cell program. However, little is known of the order or timing of such changes or of the cellular context in which they occur. Does cancer arise through the malignant transformation of normal stem cells or of committed downstream progenitors (or both)?56 Unfortunately, the “stem” in “cancer stem cell” has led to the frequent misconception that CSCs always arise from stem cells.57 On the contrary, emerging evidence suggests that although some cancers may originate in normal stem cells, committed progenitors can also be initial targets for transformation.
There has been a great deal of debate over the cellular origins of cancer. A number of conceptual arguments have been invoked to support stem cells as the cell of origin. One contention is that because self-renewal is a key property of CSCs, and stem cells already possess self-renewal capacity, they would theoretically require fewer neoplastic changes in order to become fully transformed. In contrast, despite their substantial proliferative ability, progenitors do not generally retain the self-renewal capacity of stem cells. Thus to become a CSC, a progenitor must acquire mutations that reactivate the cellular self-renewal machinery. In addition, there is greater opportunity for genetic changes to accumulate in individual, long-lived stem cells compared to more mature progenitors with a limited lifespan. If a short-lived progenitor acquires a genetic mutation that does not confer increased self-renewal, that cell is likely to die or undergo terminal differentiation before enough mutations occur for full neoplastic transformation.
On the other hand, stem cells are fewer in number and divide less frequently than progenitors; therefore, the probability that they will acquire the mutations needed for cancer development is correspondingly lower. Furthermore, some have argued that stem cells are the key to tissue regeneration and the only cells in adult tissues capable of self-renewal. Thus, they might have evolved sophisticated inhibitory machinery to keep self-renewal in check as an anticancer defense.58 If this is true, stem cells could actually be less likely to acquire mutations that affect self-renewal and proliferation or to manifest biological changes as a result of such mutations, as compared to more committed progenitors. In other words, stem cells might be under evolutionary pressure to preserve genomic integrity over promoting survival in order to prevent oncogenic transformation.
Functional Studies in Leukemia
It is problematic to make inferences regarding the cellular origins of CSCs simply by studying their surface phenotype, as disruption of normal differentiation pathways by the neoplastic process can lead to aberrant expression of lineage-associated markers. Similarly, it is difficult to draw firm conclusions by studying the lineage involvement of the neoplastic clone. For example, in chronic myeloid leukemia (CML) patients, involvement of multiple hematopoietic lineages has been taken as evidence that CML originates from a multipotent HSC, whereas lineage restriction in some AML patients has been interpreted as disease origin from a committed progenitor. However, apparent lineage restriction of the leukemic clone in AML could also result from mutations that arise in a multipotent stem cell and suppress differentiation to one or more lineages.59 The most direct way to determine whether CSCs arise from neoplastic transformation initiated in stem cells or progenitors is to test whether oncogene expression in directly isolated, functionally validated normal stem and progenitor cell populations is able to confer in vivo tumor-initiating ability.
This approach has been used to study leukemic initiation in the murine hematopoietic system, facilitated by previous detailed phenotypic and functional characterization of different classes of progenitors and HSCs. One focus of investigation has been the mixed-lineage leukemia (MLL) gene, which undergoes fusion with a wide variety of partner genes and is associated with myeloid, lymphoid, and biphenotypic acute leukemias in both children and adults. The fusion gene MLL-GAS7 induces mixed-lineage leukemias when expressed in murine HSCs or multipotent progenitors, but not in lineage-restricted progenitors.60 In contrast, MLL-ENL can initiate myeloid leukemias in both self-renewing HSCs and committed myeloid progenitors,61 although much lower numbers of transformed HSCs compared to progenitors were required for tumor initiation in vivo. MLL-AF9 is also able to generate LSCs from committed granulocyte macrophage progenitors (GMPs).45 Interestingly, when MLL-AF9 is expressed at lower, physiologic levels in a knock-in transgenic mouse model of leukemia, HSCs and CMPs, but not GMPs, from transgenic mice are able to initiate leukemia upon transplantation into wild type recipients.62 Compared to retrovirally transduced GMPs, GMPs from knock-in mice had 170-fold lower expression of MLL-AF9. These findings imply that differences in experimental transformability of HSC and progenitor populations may be related to oncogene dosage. However, the nonequivalent transformation of HSCs and committed progenitors, even by supraphysiologic levels of retrovirally driven MLL-ENL,61 points to the existence of inherent differences in susceptibility between these populations.
Taken together, these findings in the murine system indicate that MLL-expressing leukemias may be initiated in either HSCs or downstream progenitors, depending on the specific fusion partner involved. This likely depends on the ability of the fusion oncogene to reactivate a self-renewal program in committed progenitors. Indeed, the LSCs generated by MLL-AF9 expression in GMPs possessed a surface immunophenotype and global gene expression profile similar to that of normal GMPs, but also demonstrated reactivation of a subset of genes highly expressed in HSCs and associated with self-renewal.45,63 The capacity of MLL fusion genes to reactivate self-renewal machinery in progenitors confers a potent transforming ability to this group of oncogenes, as evidenced by the high frequency of leukemias in NOD/SCID mice transplanted with primitive human hematopoietic cells expressing an MLL fusion gene44 and the infrequency of additional detectable genetic changes in human acute lymphoblastic leukemias with MLL rearrangements.64
Conclusions drawn from studies in MLL leukemias may not be generalizable, as MLL leukemia appears to be a unique disease distinct from other subtypes of acute leukemia.65 However, there have been a few reports of the transforming ability of other leukemia-associated oncogenes. MOZ-TIF can initiate AML in HSCs as well as committed progenitors, while BCR-ABL1 did not increase self-renewal in vitro or initiate disease when expressed in committed progenitors, despite the induction of myeloproliferative disease in mice transplanted with BCR-ABL1-transduced whole bone marrow.43 These findings suggest that BCR-ABL1 expression in more primitive HSCs is required for disease initiation, although this was not tested directly. Nevertheless, these results are consistent with data from studies of transgenic mouse models of CML, in which BCR-ABL1 expression66 or inactivation of JunB, a transcriptional regulator of myelopoiesis, in HSCs induces a transplantable myeloproliferative disorder, whereas JunB inactivation in more committed progenitors does not.67 Interestingly, analysis of the JunB-deficient mice demonstrated expansion of the primitive HSC compartment as well as the GMP pool; however, the numbers of common myeloid progenitors and megakaryocytic-erythroid progenitors were similar to those of control animals, indicating that oncogenic changes in stem cells can have very specific effects in downstream progeny. Expansion or extended proliferation of progenitors may increase the risk of acquiring secondary cooperating events, resulting in progression to a fully transformed state. For example, transgenic mice that express reduced levels of the transcription factors PU.168 or GATA-169 are characterized by accumulation of an abnormal progenitor pool and a high propensity to develop acute leukemias, which in the PU.1 knockdown mice are frequently accompanied by additional chromosomal abnormalities. The mutant alleles in these engineered mice were expressed in every cell, thus it is impossible to determine whether reduced transcription factor levels would have equivalent transforming potential in the context of HSCs or progenitors. Patients with chronic phase CML have an expanded progenitor pool related at least in part to expression of BCR-ABL. Progression to blast crisis is associated with further expansion of GMPs that have activated β-catenin activity and increased self-renewal as demonstrated by in vitro replating assays.70
Overall, the accumulated evidence from studies in murine hematopoiesis indicates that the neoplastic changes that lead ultimately to generation of LSCs can be initiated in either normal HSCs or progenitors, depending on the ability of specific transforming events to overcome the inherent cellular barriers to transformation (Fig. 11.3). Potent oncogenes such as MLL that can reactivate self-renewal are able to transform committed progenitors, whereas less potent oncogenic changes must occur in self-renewing HSCs in order to initiate a tumorigenic program. Secondary neoplastic changes can occur within the stem cell compartment or within abnormally expanded downstream progenitor populations, ultimately resulting in the generation of LSCs. While the murine studies described above have provided significant insights, it will be important to carry out equivalent studies in the human hematopoietic system, as the processes underlying neoplastic transformation differ between mouse and man.71
Studies in Solid Tumors
Similar studies in solid tumors have been hampered by the lack of phenotypically and functionally defined stem and progenitor cell populations in most normal tissues. Recently, stem cells have been identified in murine72,73 and human74 mammary tissue and in murine prostate.75–77 Lim et al.78 proposed that luminal progenitors, rather than mammary stem cells, are the cell of origin for human basal breast cancers, which are associated with germline mutations in the tumor suppressor gene BRCA1, based on stronger similarity in gene expression profiles. Furthermore, the luminal progenitor population in premalignant breast tissue from BRCA1 mutation carriers is expanded and has increased growth potential in vitro. However, analogous to targeting of HSCs in CML, it is also possible that BRCA1 mutations target mammary stem cells and either drive development preferentially down the luminal lineage or have biological consequences only upon luminal differentiation. Transformation experiments of mammary cell subsets are needed to clarify this issue.
Despite the recent progress in characterizing normal cellular hierarchies in nonhematopoietic tissues, direct testing of the transformability of isolated stem and progenitor populations remains a challenge in most systems. Thus, researchers have turned to alternative methods to investigate the cell of origin in solid tumors. In brain cancer, genetic approaches have provided insights into the normal cell populations that might be targeted for transformation. Investigators used a transgenic mouse system that allows expression of oncogenes in a cell type-specific manner to show that selective expression of platelet-derived growth factor in nestin+ neural stem/progenitor cells generated oligodendrogliomas, whereas expression in GFAP+ astrocytes generated a mixture of oligodendrogliomas and oligoastrocytomas.79 Similarly, the combined expression of activated Ras and Akt in nestin+ but not GFAP+ cells generated high-grade glioblastomas.80 These findings suggest the existence of inherent differences in the transformability of neural cell populations but are confounded by the fact that GFAP marks neural stem cells as well as differentiated astrocytes.81 In contrast, transduction of a constitutively active endothelial growth factor receptor mutant into either neural stem/progenitor cells or cortical astrocytes from Ink4a/Arf−/− mice gave rise to high-grade gliomas with similar latency, indicating that both cell compartments are permissive for transformation.82 However, the isolated cell populations in this study were transduced only after a period of in vitro culture and were derived from age-mismatched animals, factors that can hamper direct comparisons of primitive and mature cell populations.
Using a different transgenic model, two groups found that activation of hedgehog (Hh) signaling, which is etiologically linked to medulloblastomas in mice and humans,83 in both multipotent stem/progenitor cells and unilineage granule neuron precursors (GNPs), but not Purkinje neurons, generated transplantable medulloblastomas, but no tumors of other histological types.84,85 Interestingly, Hh activation in stem/progenitor cells resulted in expansion and increased proliferation of these cells as well as committed GNPs, but caused no abnormalities in other downstream cell populations,85 suggesting that Hh activation must occur in a lineage-specific context to manifest biological effects. The generation of medulloblastomas and no other histological tumor types is likely related to specific influences of Hh activation on target cell development, because conditional inactivation of tumor suppressor genes p53, NF1, and Pten (commonly mutated in human astrocytomas) in neural stem/progenitor cells generates high-grade astrocytomas.86,87
In the intestine, genetic lineage–tracing studies in mice have recently identified adult stem cell populations,88–90 enabling studies of the cellular origin of intestinal tumors. Most human colorectal cancers are etiologically linked to mutations in the tumor suppressor gene Apc, with resultant activation of Wnt/β-catenin signaling.91 In transgenic mouse models, rapid adenoma formation was consistently seen with conditional Apc deletion or expression of a stable β-catenin mutant in stem cell populations but not in more differentiated progenitors,88,90,92 supporting a stem cell origin for intestinal tumors in the mouse. However, the findings from these transgenic models with full, constitutive β-catenin activation may not be fully applicable to human colorectal cancer, where studies have suggested that a specific degree or “dosage” of β-catenin signaling may be optimal for tumor formation,93 as has been shown for leukemia stem/progenitor cells and MLL signaling. Clarification of these issues will require studies using human cells as target populations.
It is clear from these murine studies that cancers can originate in both stem cell and progenitor compartments, depending on the oncogene(s) involved, although stem and progenitor cells might differ in their inherent susceptibility to transformation. In addition, tumors generated by specific oncogenes are often phenotypically and functionally similar regardless of the cell of origin, suggesting that tumor heterogeneity in tissues is generated in large part by the differential effects of transforming events on the developmental program of the targeted cells, rather than by differences among target cells themselves. However, the results from murine studies must be confirmed in equivalent human studies using primary, functionally defined stem and progenitor cell populations. Insight into the cellular context in which the first steps of neoplastic transformation occur will be vital not only to understand how normal developmental processes become subverted during cancer initiation and progression, but also to advance our knowledge of CSC biology and facilitate the development of novel and effective therapies to eliminate these cancer-sustaining cells in patients.
EPITHELIAL-MESENCHYMAL TRANSITION
Epithelial-mesenchymal transition (EMT) is a complex molecular and cellular program involved in embryonic development whereby epithelial cells lose their differentiated characteristics and acquire a mesenchymal phenotype. There is recent evidence that EMT induction contributes to tumor progression by conferring properties such as invasiveness, the ability to metastasize, and resistance to therapy on epithelial cancer cells.94,95 EMT induction of ras-transformed, SV40-immortalized human mammary epithelial cells (HMLEs) significantly increased their ability to generate tumors in vivo following subcutaneous injection into immune-deficient mice.96 Notably, despite having undergone an EMT, transformed HMLEs did not exhibit in vivo tumor-initiating ability when EMT-inducing signals were removed, and the cells reverted to an epithelial phenotype when cultured further in vitro.96 A number of groups have reported that EMT-inducing signals can originate from stromal cells in the tumor microenvironment.97,98 Together, these observations suggest that in some epithelial tumors, acquisition and maintenance of CSC-like properties depend on continuous signals from the microenvironment, and support the notion that metastatic cancer cells revert to an epithelial state after dissemination and resultant loss of contact with a “tumor microenvironment.” However, sustained induction of EMT might in some cases lead to irreversible, heritable epigenetic alterations that maintain the mesenchymal phenotype.99 Such changes could have therapeutic implications, as breast cancer cells induced into EMT appear to be more resistant to standard chemotherapeutic drugs compared to control cells.100 Furthermore, EMT of non-self-renewing cancer cell populations within a tumor could contribute to clonal evolution and intratumor heterogeneity of CSCs (Fig. 11.3). It should be noted, however, that the linkage between EMT and tumor initiation is still tenuous; a deeper understanding will require clonal assays, well-defined cell fractions, proof of self-renewal, and studies of primary tumors rather than cell lines.
CANCER STEM CELLS: TARGETED THERAPY
General Considerations
Implicit in the model of cancer as a hierarchical disease is the notion that CSCs are biologically distinct from the bulk cells in the tumor. Molecular pathways for survival and response to injury may be fundamentally different in these cells compared to nontumorigenic cells. Ultimately, to prevent disease relapse and achieve permanent cure, the CSCs that sustain tumor growth must be eradicated in addition to killing the bulk cells of the tumor. However, properties of CSCs, such as quiescence or expression of drug-resistance transporters, may make them difficult to eliminate using conventional cytotoxic drugs that kill the bulk tumor cells. It will be crucial to understand the unique biology of CSCs in order to develop novel treatments that effectively target these cells (Fig. 11.4).
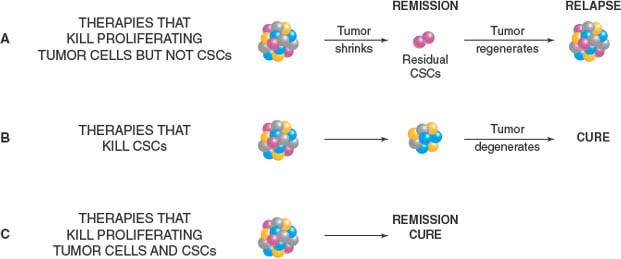
FIGURE 11.4 Development of effective anticancer therapies. A: Cancer stem cells (CSCs) are biologically distinct from bulk tumor cells and may not be effectively killed by conventional anticancer therapies due to properties such as quiescence or expression of drug-resistance transporters, leading to regrowth of the tumor and relapse after treatment. B: Ultimately, to prevent disease relapse and achieve permanent cure, the CSCs that sustain tumor growth must be eradicated. C: The most effective anticancer strategies will involve combination regimens that both reduce tumor bulk and kill CSCs, the latter likely best achieved through targeting of multiple critical pathways (see text). The development of new CSC-targeted therapies will require a greater understanding of the molecular pathways that drive tumor initiation and progression.
There are several obstacles to be overcome in the development of effective CSC-targeted therapies. Such treatments must be selective for CSCs and spare normal stem cells. There is recent evidence in AML that the pathways that regulate self-renewal in normal stem cells are not completely abolished in LSCs.30 In other words, CSCs, although transformed, likely retain aspects of normal developmental pathways. Thus, drugs that target critical processes in CSCs, such as survival or self-renewal, may prove intolerably harmful to their normal counterparts. Furthermore, normal stem or progenitor cells may in fact be more sensitive than CSCs to the effects of chemotherapy. CSCs will likely have acquired genetic or epigenetic changes that allow them to bypass normal tumor-suppressing processes such as senescence or apoptosis in response to DNA damage. Thus, treatment with agents that normally induce senescence or apoptosis may actually provide a growth advantage to CSCs.101 Ideally, effective therapies will target pathways that are necessary for CSC survival but not for the survival of normal stem cells.
Clinical testing of CSC-targeted therapies must take into account the relative infrequency of these cells within some tumors. Treatments that eradicate CSCs may not have significant effects on proliferation or apoptosis of the bulk of tumor cells102 and therefore may not cause rapid tumor regression or shrinkage.There is a significant risk that agents that selectively target CSCs will be overlooked in clinical trials if assessed simply on the basis of objective tumor response. In the evaluation of CSC-targeted therapies, delay in tumor progression would be a more relevant clinical end point. In the end, the ultimate test of the effectiveness of a CSC-targeted agent is whether relapse is prevented, an end point that requires long-term follow-up. A corollary to this concept is that evaluation of toxicity toward normal stem cells should also be measured in long-term studies, in order to properly assess effects on tissue maintenance. As tumor shrinkage is not an accurate measure of the efficacy of agents that selectively kill CSCs, clinical evaluation would be greatly aided by the development of sensitive real-time imaging modalities to detect and quantify residual CSCs in patients who undergo treatment. However, such technology is currently unavailable. As discussed above, current protocols for isolating tumor cell fractions enriched in CSCs do not yield pure populations, thus it is problematic simply to correlate therapeutic effectiveness with eradication of a phenotypically defined cell population. Ultimately, detection of residual CSCs with the ability to reinitiate tumor growth may depend on functional assays of tumorigenicity. However, such testing would be subject to the inherent limitations associated with in vivo detection of rare cells.
Resistance to Standard Therapy
The identification of CSCs in solid tumors and the development of in vivo assays to assess their tumorigenic properties have paved the way for studies to assess the impact of anticancer therapies on these cells. Radiation therapy for glioblastoma multiforme, an aggressive type of brain cancer, is transiently effective but is often followed by tumor recurrence or progression, implying that CSCs are not effectively eradicated. This notion is supported by recent evidence that ionizing-radiation treatment of glioblastoma grafts grown in mice leads to an increase in the proportion of CD133+ cells in the residual tumor population compared to unirradiated controls.101 Irradiated CD133+ cells retain the ability to form heterogeneous tumors in vivo that can be serially propagated. In vitro experiments demonstrated that this radioresistance is likely due to increased activation of DNA damage checkpoint proteins and more efficient repair of DNA damage, resulting in a lower rate of apoptosis compared to CD133− cells. Intriguingly, treatment of CD133+ cells with an inhibitor of two checkpoint kinases disrupted their radioresistance, although the ability of treated cells to initiate tumor growth in vivo was not tested. A recent study that showed lower levels of pro-oxidants and higher expression of antioxidant genes in the CSC-enriched CD44+CD24−/lowLineage− cell population from primary human breast cancers compared to nontumorigenic cells suggests that lower levels of reactive oxygen species may also contribute to the relative radioresistance of CSCs.103
There have been a number of studies that indicated that CSCs may be resistant to standard chemotherapy. Following in vitro treatment of human pancreatic cancer cells with the nucleoside analogue gemcitabine, the CD133− population underwent apoptosis and was dramatically reduced, whereas most CD133+ cells survived, resulting in nearly 40-fold enrichment of the CD133+ fraction.37 In addition, in vivo gemcitabine treatment of mice bearing pancreatic cancer cell line xenografts resulted in reduction of tumor bulk but approximately fourfold enrichment of the minority CD133+ population. These data suggest that the CSC-containing CD133+ cell population is relatively resistant to chemotherapy compared to non-CSCs. However, as discussed above, phenotypically defined populations enriched for CSCs are heterogeneous and contain non-CSCs as well. Thus, it is crucial to measure the effects of treatment on CSCs directly through functional assays. For example, Li et al.104 showed that human breast cancer biopsy samples obtained from patients treated with standard chemotherapy had increased percentages of CD44+CD24−/low cells and increased clonogenic efficiency in vitro, implying enrichment of CSCs. Importantly, posttreatment samples were twice as efficient at generating tumor xenografts when implanted into the mammary fat pads of SCID/Beige mice, confirming a functional enrichment of CSCs surviving in the treated tumors.
CD133+ human colon cancer cells are more resistant to apoptosis induced by in vitro treatment with 5-fluorouracil or oxaliplatin compared to CD133− cells.105 In vivo oxaliplatin treatment of mice bearing colon cancer xenografts led to a reduction in tumor size but an increase in the percentage of CD133+ cells, with associated resistance of CD133+ cells to apoptosis. Cotreatment of mice with oxaliplatin plus a neutralizing antibody against IL-4 resulted in delayed tumor growth that persisted after treatment was discontinued, as well as increased apoptosis of CD133+ cells, leading to enhanced killing of these cells. These findings suggest that chemoresistance of colon CSCs may be related at least in part to autocrine IL-4 signaling, possibly through up-regulation of an antiapoptotic program.105 Similarly, in vivo treatment of early passage colon cancer xenografts with cyclophosphamide led to slowing of tumor growth but enrichment of cells with a CSC phenotype and a twofold increase in CSC number measured by limiting dilution serial transplantation assays.106 ALDH1 activity catabolizes the cytotoxic metabolites of cyclophosphamide; short hairpin RNA–mediated knockdown of ALDH1 sensitized tumors to cyclophosphamide treatment in vivo and reduced the frequency of CSCs in residual tumors. Thus, identification of functional pathways such as ALDH1 that are important for CSC survival can provide not only markers for isolating these cells, but potential therapeutic targets as well. Future studies using primary human tumors tested in in vivo functional assays will be required to determine the clinical importance of CSCs in tumor response and relapse.
Targeting the Microenvironment
Angiogenesis is a critical factor in the early stages of tumor formation as well as in tumor progression and therefore represents a potential target for anticancer treatments. Regulation of angiogenesis involves a complex interplay between tumor cells and the neovasculature. Glioblastomas, for example, express high levels of vascular endothelial growth factor (VEGF).107 A functional interaction between brain CSCs and endothelial cells is supported by the close association of CD133+ brain cancer cells with vascular endothelial cells in vitro and in vivo, and more importantly by the demonstration that coinjection of primary human endothelial cells enhances tumor formation by CD133+ medulloblastoma cells in immune-deficient mice.102 Tumors initiated in mice by CD133+ cells from either primary glioblastoma biopsy specimens or xenograft cell lines are highly vascular.107 Interestingly, treatment of xenograft tumors with bevacizumab, an antibody that neutralizes VEGF, not only potently inhibits tumor growth in mice102,107 but also results in depletion of cells coexpressing CD133 and nestin, a marker of primitive neural cells, without directly affecting bulk tumor cell proliferation or death. Together, these results suggest that inhibition of brain tumor growth by antiangiogenic agents is mediated at least in part by disruption of a vascular niche required for maintenance of CSCs. For the most part, these experiments were done using brain cancer cell lines rather than freshly isolated tumor cells, but they nevertheless make a compelling case for further characterization and therapeutic targeting of the unique microenvironment of CSCs.
Ginestier et al.16,108 recently showed that human breast cancer cells expressing CXCR1, a receptor that binds the proinflammatory chemokine IL-8, are present almost exclusively within the CSC-containing ALDH1+ population. IL-8 has been implicated in tumor invasion, metastasis, and self-renewal.109,110 Treatment of orthotopically transplanted tumors in NOD/SCID mice with the CXCR1/2 inhibitor repertaxin, the standard chemotherapeutic agent docetaxel, or a combination of both drugs all resulted in impaired tumor growth. However, tumors treated with docetaxel alone showed either unchanged or increased percentage of ALDH1+ cells compared with untreated controls, whereas repertaxin treatment alone or in combination with docetaxel significantly reduced the ALDH1+ population. Importantly, upon serial transplantation, tumor cells derived from control or docetaxel-treated primary animals were able to generate tumors in secondary mice with similar efficiency, while cells from repertaxin-treated animals showed a two- to fivefold reduction in tumor growth and were only able to generate tumors at the highest injected cell dose. These findings indicate that, contrary to standard chemotherapy with docetaxel, CXCR1 blockade with repertaxin directly targets and reduces the CSC population in vivo and underscore the importance of directly assaying effects on CSCs rather than relying on traditional measures of efficacy such as reduction of tumor bulk.
Differentiation Therapy
Another approach to anticancer therapy is induction of differentiation of CSCs, with consequent associated loss of self-renewal capacity. This strategy has been highly successful in acute promyelocytic leukemia (APL), where the addition of retinoic acid to conventional chemotherapy has significantly improved survival rates. The demonstration that the growth of solid tumors is sustained by CSCs with the capacity to generate tumor heterogeneity implies that it should be similarly possible to drive the differentiation of CSCs in these cancers. However, the clinical development of differentiation-inducing agents to treat solid tumors has been limited to date. Bone morphogenic proteins (BMPs) are soluble factors that induce normal neural precursor cells to differentiate. BMP treatment of CD133+ glioblastoma cells in vitro or in immune-deficient mice results in the formation of smaller, more differentiated, less invasive tumor grafts that cannot be serially propagated in mice,111 demonstrating the therapeutic potential of differentiation-inducing agents in brain cancer. However, the differentiation response to BMP treatment may be impaired in a subset of glioblastoma CSCs due to epigenetic silencing of BMP receptor 1B (BMPR1B), a feature shared with early embryonic neural stem cells.112 Indeed, in patient-derived glioblastoma cells with reduced BMPR1B expression, BMP2 induced proliferation rather than differentiation. Transgenic expression of a constitutively active BMPR1B mutant in these cells followed by orthotopic injection into neonatal SCID mice resulted in higher expression of GFAP by tumor cells, impaired tumor growth, and improved host survival compared to unmanipulated tumor cells. BMPR1B down-regulation was found in approximately 20% of primary human glioblastomas. This elegant study not only provides insight into the role of BMPs in brain CSC biology, but highlights the existence of intertumor CSC variability, even among tumors of the same histological class. As discussed above, tumor heterogeneity is likely related in large part to differences in the underlying oncogenic changes that drive tumor growth. Thus, therapeutic approaches such as induction of differentiation may require targeting of multiple pathways and will likely need to be tailored to some degree to the biology of individual tumors. A recent unbiased RNA interference screen identified several genes whose silencing induced a “differentiation phenotype” in glioblastoma cells derived from multiple patients’ tumors.113 The strongest and most reproducible of these was TRRAP, which encodes an adaptor protein found in multiprotein/chromatin complexes. Knockdown of TRRAP in three different patient-derived glioblastoma lines followed by orthotopic injection into SCID mice resulted in impaired tumor growth, a more differentiated xenograft phenotype, and improved host survival compared to control cells. Future studies are required to determine the applicability and efficacy of these treatment approaches in the clinic.
Selected References
The full list of references for this chapter appears in the online version.
1. Bruce WR, van der Gaag H. A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature 1963;199:79.
2. Southam CM, Brunschwig A, Dizon Q. Autologous and homologous transplantation of human cancer. In: Brennan MJ, Simpson WL, eds. Biological interactions in normal and neoplastic growth. A contribution to the host-tumor problem. Boston: Little, Brown, 1962:723.
5. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukemia after transplantation into SCID mice. Nature 1994;367:645.
6. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730.
7. Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000; 14:1777.
8. Jin L, Lee EM, Ramshaw HS, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 2009;5:31.
10. Guzman ML, Rossi RM, Karnischky L, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 2005;105:4163.
12. Yilmaz OH, Valdez R, Theisen BK, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006;441:475.
14. Jin L, Hope KJ, Zhai Q, et al. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med 2006;12:1167.
15. Al Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003;100:3983.
21. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature 2004;432:396.
25. Chen R, Nishimura MC, Bumbaca SM, et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010;17:362.
28. O’Brien CA, Pollett A, Gallinger S, et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007;445:106.
29. Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature 2007;445:111.
30. Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol 2004;5:738.
37. Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007;1:313.
38. Kelly PN, Dakic A, Adams JM, et al. Tumor growth need not be driven by rare cancer stem cells. Science 2007;317:337.
40. Quintana E, Shackleton M, Sabel MS, et al. Efficient tumour formation by single human melanoma cells. Nature 2008;456:593.
42. Kennedy JA, Barabe F, Poeppl AG, et al. Comment on “Tumor growth need not be driven by rare cancer stem cells.” Science 2007;318:1722.
42a. Ishizawa K, Rasheed ZA, Karisch R, et al. Tumor-initiating cells are rare in many human tumors. Cell Stem Cell 2010;7:279.
43. Huntly BJ, Shigematsu H, Deguchi K, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004;6:587.
44. Barabe F, Kennedy JA, Hope KJ, et al. Modeling the initiation and progression of human acute leukemia in mice. Science 2007;316:600.
45. Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006;442:818.
48. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta 2010;1805:105.
54. Notta F, Mullighan CG, Wang JCY, et al. Evolution of human BCR-ALB1 lymphoblastic leukaemia-initiating cells. Nature In Press.
54a. Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 2010 doi:10.1038/nature09650.
60. So CW, Karsunky H, Passegue E, et al. MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell 2003;3:161.
61. Cozzio A, Passegue E, Ayton PM, et al. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev 2003;17:3029.
62. Chen W, Kumar AR, Hudson WA, et al. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer Cell 2008;13:432.
63. Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006;10:257.
67. Passegue E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 2004;119:431.
70. Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 2004;351:657.
78. Lim E, Vaillant F, Wu D, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 2009; 15:907.
79. Dai C, Celestino JC, Okada Y, et al. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev 2001;15:1913.
80. Holland EC, Celestino J, Dai C, et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 2000;25:55.
82. Bruggeman SW, Hulsman D, Tanger E, et al. Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell 2007;12:328.
84. Schuller U, Heine VM, Mao J, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 2008;14:123.
85. Yang ZJ, Ellis T, Markant SL, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 2008;14:135.
90. Zhu L, Gibson P, Currle DS, et al. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 2009;457:603.
92. Barker N, Ridgway RA, van Es JH, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009;457:608.
96. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704.
100. Gupta PB, Onder TT, Jiang G, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009;138:645.
101. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756.
102. Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007; 11:69.
104. Li X, Lewis MT, Huang J, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008;100:672.
105. Todaro M, Alea MP, Di Stefano AB, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007;1:389.
106. Dylla SJ, Beviglia L, Park IK, et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One 2008;3:e2428.
107. Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 2006;66:7843.
108. Ginestier C, Liu S, Diebel ME, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest 2010;120:485.
111. Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006;444:761.
112. Lee J, Son MJ, Woolard K, et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 2008;13:69.
113. Wurdak H, Zhu S, Romero A, et al. An RNAi screen identifies TRRAP as a regulator of brain tumor-initiating cell differentiation. Cell Stem Cell 2010;6:37.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


