FIGURE 25.1 A schematic diagram of the genomic organization of the gene responsible for multiple endocrine neoplasia type 1 (MEN-1), including MEN-1 germline and somatic mutations. The gene contains ten exons (the first of which remains untranslated) and extends across 9 kb. It encodes a 610 amino acid protein, menin. Mutations shown above the exons cause menin truncation; those shown below the exons cause an amino acid or codon change. All unique mutations are represented; numbers in parentheses represent multiple reports of the same mutation in apparently unrelated individuals. The green-shaded areas indicate the untranslated regions. The location of the two nuclear localization signals (NLS), at codons 479–497 and 588–608, are indicated. Missense mutations in a region of menin (amino acids 139–242, identified by blue shading) prevented interaction with the AP1 transcription factor JunD. (From ref. 6, with permission.)
Familial Hypocalciuric Hypercalcemia (FHH, MIM 145980). The calcium-sensing receptor (CASR), a critical regulator of extracellular calcium homeostasis, is a seven-transmembrane-spanning G-protein–coupled receptor, which is expressed in cells of the parathyroid gland and the kidney tubule. The discovery of the CASR was a surprise, since previously no small cation had been shown capable of acting as a ligand for a G-protein–coupled receptor.7 The human CASR, encoded by six exons of the gene located on chromosome 3q113.3-q21, is sensitive to changes in the ambient calcium concentration and when activated it inhibits parathyroid hormone (PTH) secretion and the renal reabsorption of calcium. With heterogenous inactivating mutations of the CASR gene the parathyroid cell fails to sense properly an increased serum calcium concentration, and the resulting increase in PTH secretion causes FHH, an autosomal dominant disease characterized by hypocalciuria, hypercalcemia, and parathyroid hyperplasia. It is important for clinicians to recognize this relatively mild form of familial hyperparathyroidism; although parathyroidectomy reduces the serum calcium level, it is only temporary. With inactivating mutations in both alleles of the CASR gene, neonatal severe hyperparathyroidism ([NSHPT] MIM 239200) develops with serum calcium levels in the range of 15 to 20 mg/dL. This disease represents a life-threatening emergency and urgent parathyroidectomy is indicated.8 Conversely, activating mutations of the CASR gene cause the parathyroid cells to sense that serum calcium is “elevated” when it is actually normal. There is a resulting decrease in the blood calcium level expressed as the syndrome of autosomal dominant hypoparathyroidism ([ADH] MIM 168468).9 To date approximately 115 mutations (60% inactivating and 40% activating) have been described in the CASR gene, and most are missense mutations clustered in exons 3, 4, and 7.
Hyperparathyroidism-Jaw Tumor Syndrome (HPT-JT, MIM 145001). HPT-JT is characterized by the autosomal dominant occurrence of hyperparathyroidism, ossifying fibromas of the mandible or maxilla, renal cysts or solid tumors, and uterine fibromas.10 Approximately 50 families with HPT-JT have been reported and 80% of patients have hyperparathyroidism, and in 15% of cases the parathyroid tumors are malignant. It is noteworthy that the age-related penetrance of HPT-JT is approximately 40% by age 40 in contrast to MEN-1 where the age-related penetrance is 98% by age 40.11 Members of kindreds with this disease need lifelong surveillance by physical examination and biochemical evaluation. It has even been suggested that serial ultrasound examination of the neck should be performed as parathyroid carcinoma has been reported in normocalcemic kindred members.12
The cause of HPT-JT appears to be a tumor suppressor gene, HRPT2, which is located on chromosome 1q25-q31. The HPRT2 gene consists of 17 exons and the mutations are scattered throughout the 1593 coding region, with the majority resulting in functional loss through premature truncation.13 Approximately 16 activating mutations of HRPT2, most of which are frameshift, have been identified. The HPRT2 gene encodes a 531 amino acid protein, parafibromin (named for parathyroid tumors and jaw fibromas).14 The function of parafibromin is unknown but it is thought to regulate posttranscriptional events and histone modification. There is recent evidence that parafibromin has pro-apoptotic activity, important as a tumor suppressor function.15 Germline mutations in HPRT2 have been identified in approximately half of HPT-JT families, and even in some families with MEN-2A.
Besides the evidence of germline mutations in patients with the HPT-JT somatic HRPT2 mutations have been detected in the majority of patients with sporadic parathyroid carcinoma.16,17 It is important to note that mutations in HRPT2 are rarely if ever seen in sporadic parathyroid adenomas, an important diagnostic finding distinguishing benign from malignant parathyroid tumors.15,18
Familial Isolated Hyperparathyroidism (FIHP, MIM 145000). The syndrome of FIHP is a heterogenous condition, and some kindreds thought to have this disease have been shown to have germline mutations of MEN1, CASR, or HRP-JT, suggesting that the disease represents incompletely expressed forms of MEN1, FHH, or HPT-JT.19 Over 100 families with FIHP have been reported and in most cases the causative genetic mutation is unknown, although there are convincing data that it resides on chromosome 2p13.3-14.20
The Pituitary Gland
There is no relationship between genotype and phenotype in patients with MEN-1, thus there is no way to predict the presence or behavior of pancreatic islet cell tumors or pituitary tumors. There are, however, important genetic mutations associated with other hereditary and sporadic pituitary tumors.
Carney Complex (CNC, MIM 160980). Pituitary adenomas can occur as a component of CNC, a familial disease characterized by hypersomatotropenemia, cardiac or cutaneous myxomas, spotty skin pigmentation, primary pigmented nodular adrenal disease, and testicular tumors.21 About 75% of patients exhibit subclinical increases in growth hormone (GH), insulinlike growth factor-1 (IGF-1), and prolactin. Acromegaly occurs in about 10% of patients. Genetic linkage analysis has shown two loci for CNC, one on chromosome 2p16 (CNC2), and the other on chromosome 17q22-24 (CNC1). Neither locus is associated with a specific phenotype. In more than 50% of cases the CNC has been linked to an inactivating mutation in the gene coding for the protein kinase A (PKA) type 1α subunit, PRKAR1A, at 17q24. The 2p16 locus is uncharacterized.
Familial Isolated Pituitary Adenomas (FIPA, MIM 102200). Pituitary adenomas can also occur as FIPA. Daly et al.22 evaluated 64 families with FIPA, residing in Belgium, France, Italy, and the Netherlands. Of the 138 affected family members, 55 had prolactinomas, 47 had somatotropinomas, 28 had nonsecreting adenomas, and 8 had adrenocorticotropic hormone–secreting tumors. The incidences of a homogenous (single tumor) phenotype and a heterogeneous tumors phenotype were approximately equal. Affected patients were found to have no mutations in either the MEN1 or PRKRA1A genes.
Recently, Vierimaa et al.,23 in a study of cases of low penetrance familial pituitary adenoma in Northern Finland identified loss of mutation functions in the aryl hydrocarbon receptor interacting protein (AIP) gene. AIP forms a complex with the aryl hydrocarbon receptor (AHR) and two 90-kD heat-shock proteins (HSP90). AHR is a ligand-activated transcription factor and also participates in cellular signaling pathways.
Expression profiling in two families resulted in the identification of AIP as one of the candidate genes, and a nonsense mutation (Gln14X) was also identified and found to segregate with the GH-secreting adenomas.23 Daly et al.24 studied 73 FIPA families with 156 individuals and found that 11 of them harbored 10 AIP germline mutations. Kindred members with AIP mutations, compared to those without mutations, were younger and had larger tumors. Growth hormone producing tumors predominated among family members with AIP mutations.
Multiple Endocrine Neoplasia Type 2
Clinical Features
In 1968 Steiner et al.25 described a large family with medullary thyroid carcinoma (MTC), pheochromocytomas, hyperparathyroidism, and Cushing’s syndrome. The disease was named multiple endocrine neoplasia type 2, and it is now recognized that there are three related syndromes characterized by hereditary MTC: MEN-2A (MIM 171400), MEN-2B (MIM 162300), and familial medullary thyroid carcinoma (FMTC) (MIM 155240). There is near complete penetrance but variable expressivity, as virtually all patients with MEN-2A develop MTC, approximately half develop pheochromocytomas, and 30% develop hyperparathyroidism. Less often MEN-2A is associated with cutaneous lichen amyloidosis (CLA), which develops on the upper back and serves as a precocious marker of the disorder, or Hirschsprung’s disease (HD), which is characterized by loss of ganglion cells in variable segments of the large bowel. Patients with MEN-2B develop MTC and pheochromocytomas with the same frequency as patients with MEN-2A, but rather than hyperparathyroidism, they express a generalized gastrointestinal neuromatosis and a characteristic physical appearance. Patients with FMTC develop only MTC. Of patients with hereditary MTC, 80% have MEN-2A, 15% have FMTC, and 5% have MEN-2B.
Medullary thyroid carcinoma originates from C cells, which are derived from the neural crest. The C cells have great biosynthetic capability and secrete the polypeptide hormone calcitonin (CTN) and the glycoprotein carcinoembryonic antigen (CEA). Plasma CTN serves as an excellent marker for MTC, and presently its main use is in detecting persistent or residual MTC following thyroidectomy.
The MTC is the most common cause of death in patients with MEN-2A, MEN-2B, and FMTC, and the only effective therapy is timely thyroidectomy as standard chemotherapy and external beam radiotherapy are not useful.
Molecular Genetics
In 1985 Takahashi et al.26 discovered the RET (REarranged during Transfection) protooncogene. The gene is located in the pericentromeric region of chromosome 10q11.2 and includes 21 exons. RET encodes a receptor tyrosine kinase, which is expressed in neuroendocrine cells (including thyroid C cells and adrenal medullary cells), neural cells (including parasympathetic and sympathetic ganglion cells), urogenital tract cells, and the branchial arch cells.
The RET gene has an extracellular portion, which contains four cadherin-like repeats, a calcium binding site and a cysteine-rich region, a transmembrane portion, and an intracellular portion, which contains two tyrosine kinase domains. Alternate splicing of RET produces three isoforms with either 9, 43, or 51 amino acids at the C terminus, referred to as RET9, RET43, and RET51.27,28 Mice lacking RET51 are normal, however, mice lacking RET9 have renal malformation and defects in innervation of the gut.29
RET is essential for the development, survival, and regeneration of many neuronal cells in the gut, the kidney, and the nervous system. A tripartite complex is necessary for RET signaling. One of four glial-derived neurotrophic factors (GDNF) family ligands (GFLs)—GDNF, neurturin, persephin, or artemin–binds RET in conjunction with one of four glycosylphosphatidylinositol-anchored coreceptors, designated GDNF family receptors (GFR): GFR-α1, GFR-α2, GFR-α3, or GFR-α4.30–32 The GFL-GFR complex causes dimerization of two RET molecules with activation of autophosphorylation and intracellular signaling. The C-terminal of RET contains 16 tyrosine residues, among which Y905 is a binding site for Grb7/10 adaptors, Y1015 a binding site for phospholipase Cγ, Y981 a binding site for c-Src, and Y1096 a binding site for Grb2. Tyrosine 1072 is a multidocking binding site for such proteins as SHC, SHCC, IRS1/2, FRS2, DOK1/4/5/, and Enigma. The RET receptor may activate various signaling pathways through Y1072, which thereby serves as a prerequisite for initiating transformation of RET-derived oncogenes in cell cultures and transgenic animals.33 The structure of the RET receptor complex and the molecular pathways activated when there is ligand binding or constitutive activation are shown in Figure 25.2..34
Recently, the biochemical characterization and structure of the human RET tyrosine kinase domain was reported showing that both the phosphorylated and nonphosphorylated forms adopt the same active kinase conformation necessary to bind adenosine triphosphate and substrate and have a preorganized activation loop conformation.35
In 1993 and 1994 it was shown that point mutations in the RET protooncogene cause MEN-2A, MEN-2B, and FMTC.36,37 The RET mutations are generally of two types and affect either the extracellular ligand binding site or the tyrosine kinase domain.
MEN-2A is associated with mutations involving the extracellular cysteine codons 609, 611, 618, 620 (exon 10) 630, or 634 (exon 11). The mutations associated with FMTC involve a broad range of codons, including those associated with MEN-2A, particularly 609, 618, and 620, as well as others: 768, 790, and 791 (exon 13), 804 and 844 (exon 14), or 891 (exon 15). One must be careful in making a diagnosis of FMTC, especially in small families, which span only one or two generations.
Ninety-five percent of patients with MEN-2B have a point mutation, codon M918T (exon 16), within the intracellular domain of RET. A few patients with MEN-2B have a mutation in codon A883F (exon 15). Rarely, compound heterozygous mutations in V804M with either Y806C or S904C occur in patients with a phenotype resembling MEN-2B.38 In a study of 25 patients with de novo MEN-2B, Carlson et al.39 found that the new mutation was of paternal origin in all cases. The investigators also observed a distortion of the sex ratio in both de novo MEN-2B patients and the affected offspring of MEN-2B transmitting males, suggesting a possible role for imprinting.
In patients with MEN-2A there is a clear correlation between genotype and phenotype, concerning both the pattern of clinical expression and the severity of disease.28,37,40 Over 85% of patients with MEN-2A have a 634 codon mutation, and a C634R substitution is most often associated with hyperparathyroidism.41,42 Patients with MEN-2A and CLA also have mutations at codon 634. Pheochromocytomas are associated with several codon mutations, most frequently 634, 618, 620, and 791.43 Patients with the relatively rare association of MEN-2A and Hirschsprung’s disease have mutations in exon 10, particularly codons 609, 618, and 620. Recently, the American Thyroid Association published guidelines for the diagnosis and treatment of MTC, including a list of the RET mutations so far described in patients with MEN-2A, MEN-2B, and FMTC.44
The medullary thyroid carcinoma has an early onset and a very aggressive clinical course in patients with MEN-2B, and it is moderately aggressive in patients with MEN-2A, particularly in patients with RET mutations in codons 634, 611, 618, and 620. The MTC is least aggressive in patients with FMTC. A schematic structure of the RET protooncogene with the most common codon mutations is shown in Figure 25.3..
It is important to note that approximately half of the patients with sporadic MTC have somatic M918T mutations in the MTC cells, and it has been suggested that the genotype is associated with a more aggressive phenotype, although this is controversial.45
The molecular basis for the genotype–phenotype correlations remains poorly understood; however, Iwashita et al.46 introduced specific RET codon mutations into the short and long isoforms of RET cDNA and transfected the mutants into NIH3T3 cells. High levels of transforming activity of the mutant RET genes M918T and A833F correlated with the aggressive clinical phenotypes, MEN-2B, while low levels of transforming activity of the mutant RET genes E768D, V804L, and S891A correlated with the less aggressive FMTC phenotype. A similar study evaluating not only transforming ability of RET mutants but apoptosis, anchorage-independent growth, and signaling confirmed the findings of Iwashita et al. and also demonstrated that M918T and A883F mutants significantly enhanced the suppression of apoptosis.47 It is of interest that mutations at codons 609, 618, and 620 markedly decrease the cell surface expression of RET, compared to codon 634 mutants, indicating that the former mutations impair transport of RET to the plasma membrane. One would expect this relationship considering the centrality of these RET mutations in association with MEN-2A and FMTC with Hirschsprung’s disease.

FIGURE 25.2 The RET structure and signaling network. The structure of the RET protein and the signaling network, showing docking sites and targets. RET, rearranged during transfection; GFLS, glial derived neurotrophic factor (GDNF) family ligands; ART, artemin; NTN, neurturin; PSP, persephin; GFRα, GDNF-family α receptors; Ca++, calcium ion. (From ref. 34, with permission.)
Recently, gene expression studies relating to MEN-2A and MEN-2B have been reported by two groups. Myers and Mulligan48 used cDNA microarray analysis of cell lines that expressed either the RET9 or the RET51 protein isoform to study RET-mediated gene expression patterns. They found that cells expressing RET have altered intercellular interactions correlated with increased expression of a number of cell surface molecules. The most striking expression pattern observed, however, was the up-regulation of stress response genes, specifically heat shock protein’s (HSP) 70 family members: HSPA1A, HSPA1B, and HSPA1L. Additionally, other members of several HSP families associated with stress response were up-regulated. The increased expression of HSPs, particularly of the HSP70 and HSP90 families, has been documented in breast cancer, gastrointestinal cancer, and endometrial cancer and is associated with a poor prognosis. Conversely, the expression of HSP70 levels in osteosarcomas and renal cell carcinomas is associated with an improved prognosis and a positive response to chemotherapy.49

FIGURE 25.3 A schematic diagram showing the RET tyrosine kinase receptor and ligand complex as well as genotype-phenotype correlations for patients with type 2 multiple endocrine neoplasia syndromes, including MEN-2A, MEN-2B, and familial medullary thyroid carcinoma (FMTC). The RET gene product is divided into intracellular (purple), transmembrane (orange), and intracellular domains containing tyrosine kinase activity (blue). The exons coding for each domain are shown with corresponding colors. Known RET codon mutations are listed and grouped according to the exons in which they occur. Phenotypically expressed clinical syndromes corresponding to each codon mutation are listed. (From You YNY, Lankhani V, Wells SA. The multiple endocrine neoplasia syndromes. In: Willard H, Ginsburg LS. Genomic and Personalized Medicine. 1st ed. San Diego: Academic Press; 2009:936.)
Jain et al.50 performed microarray expression analysis from pheochromocytomas and MTCs in 34 patients with MEN-2A, MEN-2B, and sporadic MTC. They found 118 probe sets that were differentially regulated in MEN-2B tumors compared to MEN-2A tumors (20 were up-regulated in MTCs from patients with MEN-2A and 98 were up-regulated in MTCs from patients with MEN-2B). Five genes were most discriminating by significance analysis microarray and correctly classified all of the cases of MTC associated with either MEN-2A or MEN-2B. The investigators found that genes involved in the process of epithelial to mesenchymal transition, many associated with the tumor growth factor β pathway, were up-regulated in MEN-2B MTCs. Also chondromodulin-1 mRNA and protein expression were localized to the malignant C cells, and its high expression correlated with the presence of skeletal deformities in MEN-2B patients.
Several groups have studied copy number imbalance in patients with MTC. One study of 37 patients with MTC, 29 with sporadic tumors, detected altered amplifications or deletions in chromosomal regions that housed genes likely to influence tumor pathogenesis.51 Additional studies showed copy number changes in 50% to 60% of MTC cases studied, most commonly in chromosomes 19p, 19q, 13q, and 11.52,53
APPLICATION OF MOLECULAR GENETICS TO CLINICAL MEDICINE
The discovery that mutations in the RET protooncogene cause MEN-2A, MEN-2B, and FMTC changed the management of patients with both sporadic and hereditary MTC and represents a paradigm for personalized genomic medicine.
Genetic Testing
DNA analysis for RET germline mutations is indicated in patients with presumably sporadic MTC, as approximately 5% of them will be found to have hereditary disease. It is imperative to screen the family members of patients with newly found germline RET mutations, since they are at risk for MTC and approximately half of them will be affected.54
Prophylactic Surgery
In patients with MEN-2A, MEN-2B, and FMTC who are shown to have inherited a mutated RET allele it is important to remove the thyroid before MTC develops or while it is still confined to the gland.55 Thus, considering patients with familial cancer syndromes, hereditary MTC fulfills each of the criteria necessary for prophylactic removal of the organ at risk: (1) near complete penetrance, such that virtually all patients who have inherited a mutated allele will develop the malignancy, (2) a highly reliable genetic test to detect family members who have inherited a mutated allele, (3) the organ at risk is expendable or its function can be easily replaced, (4) resection of the organ at risk can be performed with minimal risk, and (5) there is a sensitive tumor marker, or other indicator, to determine whether the malignancy has been prevented or cured following organ removal.
The question is when to remove the thyroid gland. Recently, four groups have suggested criteria for defining the age at which prophylactic thyroidectomy should be performed in young kindred members known to have inherited a mutated RET allele. In the first set of recommendations a consensus panel at the MEN Consortium Meeting in 2000 evaluated the relationship between specific RET codon mutations and the biological aggressiveness of hereditary MTC.40 Based on combined clinical data the panel defined three levels of thyroid cancer severity, on which to base the timing of thyroidectomy (Table 25.1). Patients with mutations in RET codons 609, 768, 790, 791, 804, or 891 (level 1) are at risk for developing MTC; however, their tumors are generally more indolent and develop at a later age than is the case in patients with other RET codon mutations. Recommendation for thyroidectomy in this group is controversial, and many clinicians base their advice for thyroidectomy on plasma calcitonin levels. In patients with mutations in RET codons 611, 618, 620, or 634 (level 2) thyroidectomy is recommended at or before 5 years of age. Patients with MEN-2B and mutations in RET codons 883 or 918 (level 3) have the most severe form of MTC, and thyroidectomy is recommended within the first 6 months of life. Subsequently, consensus panels of the British Thyroid Association, the National Comprehensive Cancer Network, and the American Thyroid Association have addressed this topic with largely confirmatory, and in some cases expanded, recommendations.44,56,57
Although these recommendations seem reasonable, it is known that there are certain factors that modify the severity of the MTC, even within individual families. For example, it has been shown in some kindreds with codon 804 RET mutations (generally associated with a nonaggressive form of MTC) that a concomitant somatic 918 codon mutation in MTC cells confers a highly malignant phenotype.58 Furthermore, it has been proposed that certain specific single-nucleotide polymorphisms (SNPs) influence the clinical behavior of the MTC, however, at present this relationship is unclear.
TABLE 25.1
RECOMMENDATIONS FOR PROPHYLACTIC THYROIDECTOMY BASED ON RET CODON MUTATION
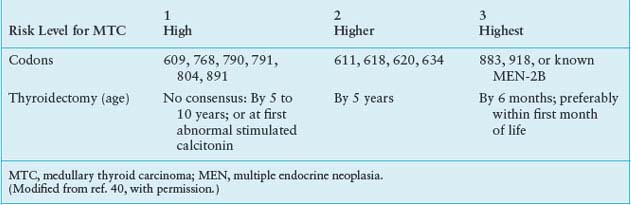
Realizing the criticalness of removing the thyroid gland while the MTC is curable, and understanding that it is impractical to establish strict guidelines for the timing of thyroidectomy based on the various RET codon mutations, clinicians should err on the side of advising thyroidectomy too early rather than too late. This approach is strengthened by the fact that once the MTC has spread beyond the thyroid gland it is virtually incurable, as no chemotherapy or radiotherapy regimen has proven effective.
SPORADIC THYROID CANCERS
Papillary Thyroid Carcinoma
Malignant tumors of the thyroid gland have a great range of biological behavior, ranging from the slow growing to highly aggressive. Over the past two decades the genetic mutations and chromosomal translocations that cause most histological types of thyroid cancer have been identified and have defined potential targets of small molecule therapeutics. The first of these observations came in 1985 with the discovery of the RET protooncogene.26 In 1987 Fusco et al.59 demonstrated in papillary thyroid carcinoma (PTC) the fusion of the C-terminal RET tyrosine kinase-encoding domain to the promoter and N-terminal portion of unrelated genes. The creation of these heterologous partners resulted in the illegitimate expression of a constitutively active chimeric oncogene termed RET/PTC. Subsequently, more than 15 molecular fusion oncogenes in PTCs have been identified, all of which differ according to the 5’-terminal region of the heterologous gene (Fig. 25.4). The most common of these chimeric oncogenes are H4(CCDC6)-RET, also known as RET/PTC1 (60% to 70%), and ELE1-RET, also known as RET/PTC3 (20% to 30%). The prevalence of RET/PTC in the thyroid cancers of children is greater than 50%, and in youngsters in Kiev and Belarus who developed PTC following exposure to radiation from the Chernobyl accident the prevalence of such rearrangements is 67% to 87%.60–62 Analysis of components of the chimeric oncogenes showed a physical proximity of the chromatin distribution of follicular epithelial cells, supporting radiation as a cause of the induced fusion.63 Similar RET/PTC rearrangements also occur in Hürthle cell carcinomas and trabecular adenomas, but they have not been described in patients with follicular carcinoma or anaplastic carcinomas.
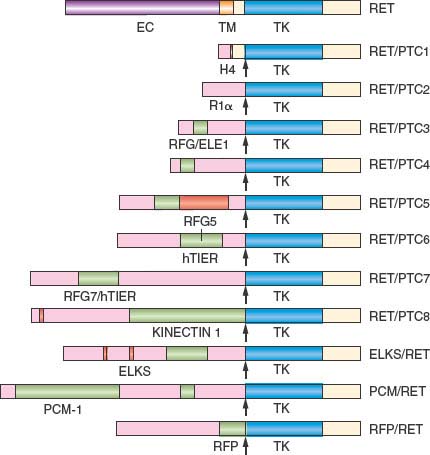
FIGURE 25.4 A schematic showing chimeric forms of the RET receptor formed by the joining of the C-terminus of RET to the promoter and N-terminus of unrelated genes. These chimeric oncogenes result in constitutive activation of RET and have been identified in patients with papillary thyroid carcinoma. EC, extracellular domain of RET; TM, transmembrane domain of RET; TK, tyrosine kinase domain of RET. H4, R1α, RFG/ELE1, RFG5, hTIF1, RFG7/hTIFR, KINECTIN1, ELKS, PCM-1, RFP represent the promoter and N-terminus of unrelated genes. (Modified from Santoro M, Melillo RM, Carlomagno F, Fusco A, Vecchio G. Molecular mechanisms of RET activation in human cancer. Ann N Y Acad Sci 2002;963:116, with permission.)
The reported incidence of these hybrid genes in sporadic PTCs varies widely, from less than 5% to almost 70%, depending on tumor heterogeneity, geographic location, and the techniques used to detect RET/PTC rearrangements. In a study of 65 papillary thyroid carcinomas, five techniques (standard-sensitivity reverse transcription-polymerase chain reaction [RT-PCR], high-sensitivity RT-PCR, real-time LightCycler RT-PCR, Southern blot analysis, and fluorescence in situ hybridization) were used to detect RET/PTC1 and RET/PTC3 rearrangements. Three patterns of detection were evident. When a significant proportion of tumor cells (35% to 86%) contained RET/PTC rearrangements (clonal pattern), the translocations were detected by all five techniques. When 17% to 24% of tumor cells (subclonal), or less than 9% of tumor cells (nonclonal), contained RET/PTC rearrangements, less than five techniques were able to detect the translocations. Also, in contrast to clonal tumors, where neither BRAF nor ras mutations were identified, such mutations were found in 40% to 60% of subclonal or nonclonal tumors. RET/PTC oncogenes, found in PTC, like the RET mutations in the MEN-2 syndromes, potentiate the intrinsic tyrosine kinase activity of RET and thereby the downstream signaling events. There does not appear to be any relationship between RET/PTC rearrangements and the clinical behavior of PTC.
A much less frequent chromosomal rearrangement associated with PTC involves the neurotrophic receptor–tyrosine kinase NTRK1 (also known as TRK and TRKA). Chimeric oncogenes result from the fusion of NTRK1 and various activating genes: TRK, which contains sequences from the TPM3 gene on chromosome 1q22-23; TRK-T1 and TRK-T2, which contain different sequences of the TPR gene on chromosome 1q25; and TRK-T3, which combines with sequences of TFG on chromosome 3q11-12.64
The most common mutation in PTC is the BRAFT17699A mutation (V600E), which occurs in 35% to 65% of tumors.65 There is virtually no overlap between the presence of BRAF mutations and RET/PTC gene rearrangements. Compared to the RET/PTC rearrangements, the BRAF mutations are reportedly associated with a more aggressive form of papillary carcinomas, characterized by extrathyroidal invasion, lymph node metastases, advanced tumor stage, and tumor recurrence. However, this point is controversial.
Follicular Thyroid Carcinoma
The peroxisome proliferators-activated receptor-γ (PPAR-γ), a member of the steroid-nuclear hormone receptor superfamily, is encoded by PPARG located on 3p25. Rearrangements involving the thyroid specific transcription factor, paired-box gene 8 (PAX8) with the peroxisome proliferators-activated receptor-γ (PPAR-γ) were first identified in follicular thyroid carcinoma (FTC) as a cytogenetically detectable translocation t(2,3)(q13; p25).66 The PAX8–PPARγ appears to be confined to atypical follicular adenomas and FTC and has not been detected in PTC or either poorly differentiated or undifferentiated (anaplastic) thyroid carcinomas. In an evaluation of 88 conventional follicular and Hürthle cell tumors analyzed for ras mutations and PAX8-PPARγ rearrangements, 49% of FTCs had ras mutations, 36% had PAX8-PPARγ rearrangements, and one had both. Ras mutations occurred in almost half of the follicular adenomas, and PAX8-PPARγ translocations were present in only 4%. Overt tumor invasiveness was associated with PAX8-PPARγ translocations and not ras mutations. PAX8-PPARγ rearrangements or ras mutations were found infrequently in Hürthle cell tumors.67
The H-, K-, and N-ras protooncogenes belong to the superfamily of membrane-associated GTP-binding proteins, which play an important role in the transduction of mitogenic signals from growth factor receptors on the cell surface. Activated ras phosphorylates Raf, which ultimately leads to activation of mitogen-activated protein kinases. Although specific patterns of ras mutations appear to be the rule in other tumors (H-ras in bladder cancer, K-ras in colon and pancreatic cancer, and N-ras in hematologic malignancies), mutations of all three ras genes have been reported in thyroid tumors.68 Most often mutations of codon 61 of H-ras or N-ras are found in thyroid neoplasms, although the incidence of the mutations varies widely, perhaps due to the various techniques used by different investigators. Garcia-Rostand et al.69 used PCR and single-strand conformational analysis to detect H-, K-, or N-ras mutations in 125 thyroid carcinomas. Mutations were present in 8.2% of 49 well-differentiated carcinomas (approximate equal frequencies in PTC and FTC), and in 50% to 55% of patients with poorly differentiated or anaplastic thyroid carcinomas (ATC). Furthermore, the mortality rate was two- to threefold higher in patients whose thyroid tumors had ras mutations, compared with those absent ras mutations.
Anaplastic thyroid carcinoma accounts for less than 2% of all thyroid cancers, but causes half of all thyroid cancer deaths. The tumor arises from follicular cells, and there is good evidence that it is a continuum of PTC or FTC. The tumors are relatively insensitive to standard chemotherapeutic regimens and are not responsive to external beam radiotherapy. The tumors are characterized by substantial chromosomal instability, characterized by BRAF or ras point mutations, which occur also in well-differentiated thyroid carcinomas and β-catenin (CTNNB1) and p53 mutations, which occur in overt ATC but not in PTC or FTC. Mutations of p53 have been described in over half of ATCs and represent a potential therapeutic target. The serine/threonine kinases of the aurora family play a central role in the regulation of the cell cycle, and the expression of Aurora B is 10- to 20-fold higher in ATC compared to normal thyroid tissue and well-differentiated thyroid carcinoma.70 A summation of the genetic mutations of thyroid cancers derived from follicular cells is shown in Table 25.2.
TABLE 25.2
GENETIC MUTATIONS ASSOCIATED WITH THYROID CANCERS DERIVED FROM FOLLICULAR CELLS

FUTURE DIRECTIONS
Advances in molecular genetics have elucidated the oncogenic event(s) of many solid tumors, and this is nowhere more evident than in endocrine tumors. The discovery of the mutations causing MEN-1 and MEN-2 has been important because it has led to an understanding of the pathogenesis, not only of the component hereditary tumors but their sporadic counterparts as well. These discoveries have already been of great benefit in the diagnosis and treatment of patients with endocrine tumors, as evidenced most clearly in families with hereditary MTC.
Another therapeutic benefit of the molecular research has been the identification of molecular targets for small molecule therapy. Already, clinical trials are under way to evaluate agents that have shown promise in preclinical studies, and some have shown significant activity.
Selected References
The full list of references for this chapter appears in the online version.
1. Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med 1954;16:363.
2. Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997;276:404.
4. Agarwal SK, Debelenko LV, Kester MB, et al/ Analysis of recurrent germline mutations in the MEN1 gene encountered in apparently unrelated families. Hum Mutat 1998;12:75.
5. Owens M, Ellard S, Vaidya B. Analysis of gross deletions in the MEN1 gene in patients with multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 2008;68:350.
6. Schussheim DH, Skarulis MC, Agarwal SK, et al. Multiple endocrine neoplasia type 1: new clinical and basic findings. Trends Endocrinol Metab 2001;12:173.
7. Brown EM, Gamba G, Riccardi D, et al. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature 1993;366:575.
8. Pollak MR, Brown EM, Chou YH, et al. Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 1993;75:1297.
10. Jackson CE, Norum RA, Boyd SB, et al. Hereditary hyperparathyroidism and multiple ossifying jaw fibromas: a clinically and genetically distinct syndrome. Surgery 1990;108:1006; discussion 1012.
12. Guarnieri V, Scillitani A, Muscarella LA, et al. Diagnosis of parathyroid tumors in familial isolated hyperparathyroidism with HRPT2 mutation: implications for cancer surveillance. J Clin Endocrinol Metab 2006;91:2827.
13. Bradley KJ, Cavaco BM, Bowl MR, et al. Parafibromin mutations in hereditary hyperparathyroidism syndromes and parathyroid tumours. Clin Endocrinol (Oxf) 2006;64:299.
16. Howell VM, Haven CJ, Kahnoski K, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet 2003;40:657.
17. Shattuck TM, Valimaki S, Obara T, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med 2003;349:1722.
19. Warner J, Epstein M, Sweet A, et al. Genetic testing in familial isolated hyperparathyroidism: unexpected results and their implications. J Med Genet 2004;41:155.
20. Warner JV, Nyholt DR, Busfield F, et al. Familial isolated hyperparathyroidism is linked to a 1.7 Mb region on chromosome 2p13.3-14. J Med Genet 2006;43:e12.
21. Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985;64:270.
22. Daly AF, Jaffrain-Rea ML, Ciccarelli A, et al. Clinical characterization of familial isolated pituitary adenomas. J Clin Endocrinol Metab 2006;91:3316.
23. Vierimaa O, Georgitsi M, Lehtonen R, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006;312:1228.
24. Daly AF, Vanbellinghen JF, Khoo SK, et al. Aryl hydrocarbon receptor interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. J Clin Endocrinol Metab 2007;92(5):1891.
25. Steiner AL, Goodman AD, Powers SR. Study of a kindred with pheochromocytoma, medullary thyroid carcinoma, hyperparathyroidism and Cushing’s disease: multiple endocrine neoplasia, type 2. Medicine (Baltimore) 1968; 47:371.
26. Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 1985;42:581.
27. Tahira T, Ishizaka Y, Itoh F, Sugimura T, Nagao M. Characterization of ret proto-oncogene mRNAs encoding two isoforms of the protein product in a human neuroblastoma cell line. Oncogene 1990;5:97.
28. Myers SM, Eng C, Ponder BA, Mulligan LM. Characterization of RET proto-oncogene 3’ splicing variants and polyadenylation sites: a novel C-terminus for RET. Oncogene 1995;11:2039.
29. de Graaff E, Srinivas S, Kilkenny C, et al. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev 2001;15:2433.
33. Ichihara M, Murakumo Y, Takahashi M. RET and neuroendocrine tumors. Cancer Lett 2004;204:197.
34. Wells SA Jr, Santoro M. Targeting the RET pathway in thyroid cancer. Clin Cancer Res 2009;15:7119.
35. Knowles PP, Murray-Rust J, Kjaer S, et al. Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem 2006;281:33577.
36. Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN2A and FMTC. Hum Mol Genet 1993;2:851.
37. Mulligan LM, Kwok JB, Healey CS, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993;363:458.
38. Miyauchi A, Futami H, Hai N, et al. Two germline missense mutations at codons 804 and 806 of the RET proto-oncogene in the same allele in a patient with multiple endocrine neoplasia type 2B without codon 918 mutation. Jpn J Cancer Res 1999;90:1.
39. Carlson KM, Bracamontes J, Jackson CE, et al. Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am J Hum Genet 1994;55:1076.
40. Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001;86:5658.
41. Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 1996;276:1575.
42. Mulligan LM, Eng C, Healey CS, et al. Specific mutations of the RET proto-oncogene are related to disease phenotype in MEN 2A and FMTC. Nat Genet 1994;6:70.
43. Machens A, Brauckhoff M, Holzhausen HJ, et al. Codon-specific development of pheochromocytoma in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab 2005; 90:3999.
44. Kloos RT, Eng C, Evans DB, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 2009;19:565.
45. Eng C, Mulligan LM, Smith DP, et al. Mutation of the RET protooncogene in sporadic medullary thyroid carcinoma. Genes Chromosomes Cancer 1995;12:209.
46. Iwashita T, Kato M, Murakami H, et al. Biological and biochemical properties of Ret with kinase domain mutations identified in multiple endocrine neoplasia type 2B and familial medullary thyroid carcinoma. Oncogene 1999;18:3919.
47. Mise N, Drosten M, Racek T, Tannapfel A, Putzer BM. Evaluation of potential mechanisms underlying genotype-phenotype correlations in multiple endocrine neoplasia type 2. Oncogene 2006;25:6637.
48. Myers SM, Mulligan LM. The RET receptor is linked to stress response pathways. Cancer Res 2004;64:4453.
49. Jaattela M. Heat shock proteins as cellular lifeguards. Ann Med 1999;31:261.
50. Jain S, Watson MA, DeBenedetti MK, et al. Expression profiles provide insights into early malignant potential and skeletal abnormalities in multiple endocrine neoplasia type 2B syndrome tumors. Cancer Res 2004;64:3907.
54. Elisei R, Romei C, Cosci B, et al. RET genetic screening in patients with medullary thyroid cancer and their relatives: experience with 807 individuals at one center. J Clin Endocrinol Metab 2007;92:4725.
55. Skinner MA, Moley JA, Dilley WG, et al. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med 2005;353:1105.
56. Sherman SI, Angelos P, Ball DW, et al. Thyroid carcinoma. J Natl Compr Canc Netw 2007;5:568.
57. Kendall-Taylor P. Guidelines for the management of thyroid cancer. Clin Endocrinol (Oxf) 2003;58:400.
59. Fusco A, Grieco M, Santoro M, et al. A new oncogene in human thyroid papillary carcinomas and their lymph-nodal metastases. Nature 1987;328:170.
60. Klugbauer S, Lengfelder E, Demidchik EP, Rabes HM. High prevalence of RET rearrangement in thyroid tumors of children from Belarus after the Chernobyl reactor accident. Oncogene 1995;11:2459.
61. Nikiforov YE, Rowland JM, Bove KE, Monforte-Munoz H, Fagin JA. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res 1997;57:1690.
63. Nikiforova MN, Stringer JR, Blough R, et al. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science 2000;290:138.
65. Kimura ET, Nikiforova MN, Zhu Z, et al. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 2003;63:1454.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


