FIGURE 9.1 Circulating bone marrow–derived cell populations that stimulate or amplify tumor angiogenesis. The various hematopoietic (CD45-positive) cell types appear to have a perivascular location with respect to the tumor neovasculature, whereas the CD45-negative endothelial progenitor cells can become incorporated into the lumen of a growing blood vessel and differentiate into mature endothelial cells. In recent preclinical studies, neutrophils have also been shown to contribute to the induction of tumor angiogenesis. F4/80 is a pan macrophage cell-surface marker. CXCR4, CXC chemokine receptor 4; RBCCs, recruited bone marrow–derived circulating cells; VE-cad, vascular endothelial-cell cadherin (an adhesion molecule); VEGFR, vascular endothelial growth factor receptor. (From ref. 4, with permission.)
The next step involves the directed locomotion/migration of endothelial cells from the parental venule toward the angiogenic stimulus emanating from the tumor mass. This is followed by division of endothelial cells that, in concert with migration, lengthen the “stalk” of the endothelial cell sprout. Subsequently, lumen formation takes place with completion of capillary sprouts and loops, and the envelopment of nascent capillaries with new basement membrane structures along with recruitment of perivascular support cells, especially pericytes. Critical in this process are specialized endothelial cells at the ends of growing capillaries called tip cells, which fuse with other tip cells to create a fused (linked) network of new capillaries.10 This sequence of events is thought to be quite similar to the formation of new blood vessel capillaries that occurs in developing embryos; however, the structure/morphology and function of many tumor-associated blood vessels can be highly irregular, heterogeneous, and functionally abnormal.6
Although this abbreviated description of sprouting angiogenesis is the most common view of angiogenesis, over the past 5 to 10 years, modifications or alternative views of angiogenesis have emerged. The mechanisms of angiogenesis may be organ- and/or tumor-specific. For example, in vascular-rich organs such as the brain, co-option may play an important role in providing a nutrient blood supply to the growing tumor.11 It has been hypothesized that parts of the vessel wall in tumors such as ocular melanoma or glioblastoma may be composed or melanoma cells, either in part (mosaic vessels)12 or full13 (“vascular mimicry”). Also noteworthy are the large number and diversity of molecular changes detected in endothelial cells in tumor blood vessels during angiogenesis, many of which suggest possible new targets for development of antiangiogenic drugs.14,15 Some of these molecular changes might be related to the recently reported genetic and cytogenetic abnormalities detected in endothelial cells isolated from the tumor vasculature16 or by endothelial cell uptake of tumor cell–derived membrane vesicles.17
PERICYTES
Pericytes (our definition of pericytes is a single layer of periendothelial smooth muscle cells) modulate endothelial cell function, and are critical for the development of a mature vascular network. Pericytes regulate vascular function, including vessel diameter (and thus blood flow) and vascular permeability.18 Pericytes also provide mechanical support and stability to the vessel wall and maintain endothelial cell survival through direct cell-cell contact and paracrine circuits.18,19
The role of pericytes within the tumor vasculature is currently an intense area of study. The degree of pericyte coverage of endothelial cells in human tumors is controversial and discrepancies among studies may be because a single marker is not sufficient to examine pericyte presence and morphology. Markers such as alpha-smooth muscle actin, desmin, NG2, and RGS5 are commonly used, and confocal imaging is necessary to observe the true relationship of pericytes to endothelial cells.
Because of the role of pericytes in mediating endothelial cell survival,19 these cells have emerged as an important therapeutic target for antiangiogenic therapy. Studies of antiangiogenic agents targeting endothelial cell survival have demonstrated that such drugs result in increased apoptosis in endothelial cells that are not associated with pericytes, leading to a relative increase in the proportion of vessels with pericyte coverage.20 These data have led to the hypothesis that pericytes mediate resistance to antiangiogenic therapy. If this hypothesis is correct, targeting both endothelial cells and pericytes will increase the efficacy of antiangiogenic therapy, and there is some evidence in support of this hypothesis in preclinical studies.21,22 However, there is currently some growing doubt about the clinical impact of targeting pericytes using drugs such as multitargeting tyrosine kinase inhibitors (TKIs), for example, sunitinib, sorafenib, and pazopanib, which target platelet-derived growth factor (PDGF) receptors (which are expressed by pericytes) in addition to vascular endothelial growth factor receptors (VEGFRs). Thus, the therapeutic impact of such drugs does not appear to be significantly greater compared with specific anti-VEGF antibodies with the exception, currently, of hepatocellular carcinoma. Moreover, there is some limited preclinical evidence that inhibition of pericyte function and attachment to endothelial cells may actually facilitate metastasis by allowing tumor cell intravasation and extravasation.23 Still others have shown that inhibition of pericyte stimulatory factors may actually increase tumor growth, as pericytes may induce endothelial cell quiescence. Pericyte biology remains an important area of research that needs to be investigated more thoroughly.
DYSFUNCTIONAL NATURE OF THE TUMOR VASCULATURE
Although tumors possess the means to recruit and develop a new vascular network, this is not to suggest, as already mentioned, that such blood vessels are normal in either structure or function. Indeed, the characteristics of the vasculature in solid tumors are associated with a number of prominent abnormalities, the consequences of which have been hypothesized to have a significant impact on tumor growth, progression, and response to various anticancer therapies. For example, the structural and morphologic abnormalities include excessively dilated blood vessels, other vessels with areas containing absent or abnormal basement membranes, or having extreme corkscrewlike tortuosities, a relative lack of supporting perivascular cellular elements such as pericytes, or abnormalities in the pericyte population, and excessive vascular leakiness.24,25 These abnormalities can be quite variable within a solid tumor mass, and such heterogeneity can also extend to the relative density of blood vessels, which can be quite high in certain areas, and low in others.
As a result of all of these features, blood flow and perfusion within tumors can be highly heterogeneous and often sluggish, with some areas therefore being deprived of oxygen and nutrients leading to adjacent areas of elevated hypoxia. This may account for slow growth of tumors in some regions and more rapid growth in others. In addition, the marked leakiness/hyperpermeability of the tumor vasculature can lead to a marked extravasation of high-molecular-weight plasma proteins and fluid into the extracellular microenvironment within tumors, which can lead to elevated interstitial fluid pressures.24 It has been hypothesized that this can limit or retard the diffusion of anticancer drugs, especially antibodies or gene therapy vectors, and immune effector cells from the blood through the interstitium of the tumors.24 Thus, given their nature, tumor blood vessels, while necessary for progressive tumor growth and hematogenous metastasis, may also actually limit the efficacy of a broad and diverse array of anticancer drugs and treatments, including chemotherapy, and oxygen required for optimal efficacy of radiation therapy.24
MOLECULAR MEDIATORS OF TUMOR ANGIOGENESIS: ANGIOGENIC STIMULATORS AND THEIR RECEPTORS
Several diverse families of growth factors (angiogenic factors) are now known to stimulate/mediate tumor angiogenesis. Some, like VEGF,26 are primary, direct-acting factors that bind to cognate receptors that are primarily expressed on endothelial cells, especially when they are “activated.” Other factors are likely secondary in nature, that is, indirect acting. In other words, they stimulate expression of one or more of the primary proangiogenic growth factors or recruit cells to sites of angiogenesis that amplify the angiogenic process. Included in this group are such molecules as transforming growth factor beta (TGF-β), TGF-α, hepatocyte growth factor, inflammatory cytokines such as interleukin-6 and interleukin-8, cytokines such as granulocyte-colony stimulating factor, chemokines such as stromal-derived factor-1, and sex hormones such as estrogens and androgens.4 PDGFs have also been implicated as mediators of angiogenesis, for the most part through their effects on PDGF receptor-expressing pericytes, as previously mentioned. However, it is the primary, direct-acting factors, foremost among them VEGF,26 that are considered to be the principal driving forces in stimulating both physiologic and pathologic angiogenesis, including tumor angiogenesis, in most cases.
Direct-acting, primary proangiogenic growth factors include the VEGF family and their cognate receptor tyrosine kinases,26 the angiopoietins, especially angiopoietin-1 and -2 (ang-1/ang-2), and their cognate tyrosine kinase receptors, in particular tie-2 (see later discussion), and the Notch signaling receptor (specifically Notch 4) and its family of ligands, such as Deltalike ligand 4 (DLL4) and Jaggeds (see later discussion). All three systems have in common a high (but not absolute) degree of specificity for endothelial cells, and in particular activated endothelial cells associated with neovascularization. Another receptor tyrosine kinase-ligand system involved in angiogenesis is Eph receptor ephrin-B2.27 Ephrin-B2 is a transmembrane ligand that is involved in “bidirectional signaling” whereby Eph receptors help regulate endothelial tip cell guidance in the sprouting and branching of new blood vessel capillaries. Ephrin-B2 mediates its effects, at least in part, by regulating VEGFR-2 function.27
Discovery of the VEGF family and their receptors (Fig. 9.2) represented a profound turning point in the field of tumor angiogenesis research and the development of antiangiogenic drugs.26 Prior to the first published reports of VEGF, which was initially called vascular permeability factor,28 basic fibroblast growth factor (bFG)F was considered to be the central mediator of angiogenesis, and was the first molecular mediator of angiogenesis to be identified.3 However, bFGF lacks a signal sequence for cellular secretion, and therapeutic blockade of bFGF using antibodies did not cause consistent antitumor results, observations that raised doubts about a predominant role for bFGF in tumor angiogenesis.
VEGF was discovered in 1989 and reported to be a highly specific and potent mitogen for vascular endothelial cells.26,29 When the genes for vascular permeability factor and VEGF were sequenced, it was realized they were the same molecule.26 The vascular permeability function of VEGF is extremely potent (50,000-fold that of histamine) and probably accounts for much of the leakiness of the tumor vasculature. It is possible that enhanced permeability may be due to intercellular gaps between endothelial cells, decreased pericyte coverage (as a second barrier to permeability), and/or specialized endothelial cell organelles called vesiculovacuolar organelles,30 transmembrane vacuoles that can form channels leading to extravasation of fluid and proteins. VEGF (VEGF-A) is the prototypical member of a family of ligands with ∼40–80% homology: VEGF-A (also called simply VEGF), VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF) (Fig. 9.2). VEGF-A (hereafter called VEGF) actually exists in a number of variant isoforms based on RNA splicing. In humans, the most common splice variants are VEGF121, VEGF165, VEGF189, and VEGF206 (whereby the number denotes the number of amino acids in the mature protein). VEGF121, the shortest isoform, is freely circulating, whereas VEGF189 and VEGF206 are strongly bound to heparin sulphate containing glycoproteins and thus remain cell-bound or sequestered in the extracellular matrix where they remain biologically inactive until mobilized by specific proteases. VEGF165 has a heparin-binding sequence but can also freely circulate. Thus, VEGF121 and VEGF165 are generally considered to be the main VEGF family members that drive tumor angiogenesis. VEGF121 and VEGF165 bind to two tyrosine kinase receptors expressed by endothelial cells. These are known as VEGFR-1 (flt-1) and VEGFR-2 or KDR in humans (kinase insert domain receptor; with flk-1 being the KDR homolog in mice).26 The major signaling receptor is VEGFR-2. In contrast, VEGFR-1 signals only weakly, after VEGF binding, despite the fact that it can bind VEGF with tenfold greater affinity compared with VEGFR-2. A naturally occurring soluble form of VEGFR-1 is thought to serve as a negative regulator in physiologic angiogenesis. In addition, neuropilins (e.g., neuropilin-1 and neuropilin-2), which can bind class 3 semaphorins involved in axon guidance, can also bind the larger VEGF isoforms, as VEGF121 lacks the domain that binds to neuropilin.31 Neuropilin likely contributes to angiogenesis by serving as a coreceptor to VEGF and enhancing binding of VEGF-A to VEGFR-2.32 Antibodies that target both VEGF and neuropilin-1 yield better antiangiogenic responses than targeting a single protein.33
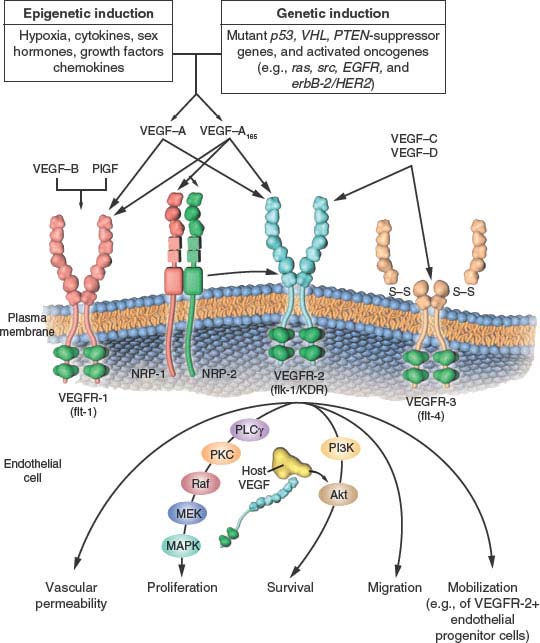
FIGURE 9.2 The family of vascular endothelial growth factor (VEGF) molecules and receptors. The major mediator of tumor angiogenesis is vascular endothelial growth factor A (VEGF-A, also called VEGF), specifically the circulating isoforms of VEGF, VEGF121 and VEGF165. These isoforms signal through VEGF receptor 2 (VEGFR-2), the major VEGF signaling receptor that mediates sprouting angiogenesis (called kinase-insert domain–containing receptor [KDR] in humans and fetal liver kinase 1 [flk-1] in mice). The role of VEGFR-1 in sprouting angiogenesis is much less clear. VEGF is expressed in most types of human cancer, and increased expression in tumors is often associated with a less favorable prognosis. Induction of or an increase in VEGF expression in tumors can be caused by numerous environmental (epigenetic) factors such as hypoxia, low pH, inflammatory cytokines (e.g., interleukin-6), growth factors (e.g., basic fibroblast growth factor), sex hormones (both androgens and estrogens), and chemokines (e.g., stromal cell–derived factor 1). Other causes include genetic inductive changes such as activation of numerous different oncogenes or loss or mutational inactivation of a variety of tumor suppressor genes. The binding of VEGF to VEGFR-2 leads to a cascade of different signaling pathways, two examples of which are shown, resulting in the up-regulation of genes involved in mediating the proliferation and migration of endothelial cells and promoting their survival and vascular permeability. For example, the binding of VEGF to VEGFR-2 leads to dimerization of the receptor, followed by intracellular activation of the phospholipase C gamma–protein kinase C–Raf kinase–MEK–mitogen-activated protein kinase (MAPK) pathway and subsequent initiation of DNA synthesis and cell growth, whereas activation of the phosphatidylinositol 3’–kinase (PI3K)–Akt pathway leads to increased endothelial cell survival. Activation of src can lead to actin cytoskeleton changes and induction of cell migration. VEGF receptors are located on the endothelial cell surface; however, intracellular (“intracrine”)-signaling VEGF receptors (VEGFR-2) may be present as well, and they are involved in promoting the survival of endothelial cells. The detailed structure of the intracellular VEGFR-2 in endothelial cells is not yet known, but it is shown as the full-length receptor that is normally bound to the cell surface. Binding of VEGF-C to VEGFR-3 mediates lymphangiogenesis. VEGF165 can bind to neuropilin (NRP) receptors, which can act as coreceptors with VEGFR-2 (horizontal arrow) to regulate angiogenesis. EGFR, epidermal growth factor receptor; flt-1, fms-like tyrosine kinase 1; PlGF, placental growth factor; PTEN, phosphatase and tensin homologue; S–S disulfide bond; VHL, von Hippel–Lindau. (From ref. 4, with permission.)
Binding of VEGF to up-regulated endothelial cell VEGFR-2 sets in motion a unique intracellular signaling cascade.34 Various investigators have identified autophosphorylation on tyrosine residues in VEGFR-2, including residues 951, 1054, 1059, 1175, and 1214. Phosphorylation of Y1175 leads to activation of phospholipase C gamma, that in turn stimulates the protein kinase C (PKC) pathway leading to inositol trisphosphate generation and calcium mobilization. In addition, this pathway, via PKCβ, stimulates the c-Raf-MEK-MAP-kinase cascade.
Another member of the VEGF family, PlGF, binds to VEGFR-1, but not VEGFR-2, and there may be circumstances where it contributes to tumor angiogenesis. Interestingly, heterodimers of VEGF-A/PlGF may prevent angiogenesis by limiting VEGF-A signaling.35 VEGF appears to be a key mediator of embryonic angiogenesis, as well as both physiologic and pathologic forms of angiogenesis in the adult. A landmark discovery in this regard was the finding that disruption and inactivation of only one of the two VEGF alleles leads to embryonic lethality associated with marked developmental abnormalities of the vasculature (“haploininsufficiency”).36,37 Homozygous disruption of flk-1/VEGFR-2 or VEGFR-1 also leads to embryonic lethality accompanied by prominent vascular defects.26
There are at least four proposed roles by which VEGF is thought to promote tumor angiogenesis26: it can stimulate endothelial division, induce locomotion/migration, enhance endothelial cell survival38 by up-regulating various inhibitors of apoptosis,39 and mobilize endothelial progenitor cells from the bone marrow to sites of angiogenesis.4 In addition, the permeability enhancing effects of VEGF might also stimulate tumor angiogenesis by causing extravasation of large molecular proteins such as fibrinogen, that can be crosslinked to form a fibrin gel in the extracellular milieu of tumors serving as a matrix for endothelial cell migration and blood vessel formation.40 VEGF also has secondary effects including up-regulation of second messengers such as nitric oxide.
VEGF is expressed by most, if not all, human (and animal) cancers, often (but not always) at much higher levels than in corresponding normal tissues. Moreover, there are many reports showing elevated VEGF is an unfavorable prognostic marker.26 The ubiquitous and elevated expression of VEGF in both human and animal tumors is likely the consequence of many factors that are commonly associated with tumors, as shown in Figure 9.2.. Among the most important is hypoxia, a prominent feature associated with the Gompertzian growth of solid tumors. Hypoxia can stabilize and hence up-regulate the levels of the hypoxia-inducible transcription factor called HIF1α, which in turn regulates hundreds of genes, among the most important of which is VEGF.41 In addition, a broad spectrum of oncogenes (e.g., ras, src, Her family members), and tumor suppressor genes, when they become mutated/inactivated or deleted, including p53, PTEN, and VHL, result in elevated VEGF expression.4
A second major growth factor signaling system that is known to be a major regulator of angiogenesis, especially for the later vessel maturation and stabilization stages, is the angiopoietin/tie-2 signaling pathway.42 There are a number of members in the family including angiopoietins-1-4 (Ang) with Ang-1 and -2 being the best characterized.43 Both of the latter bind to a highly specific endothelial cell-associated receptor tyrosine kinase, tie-2. Binding of Ang-1 to tie-2 causes an agonist effect whereas binding of Ang-2 is antagonistic. However, “pharmacologic levels” of Ang-2 may also serve as an agonist, which makes this system somewhat more complex and difficult to study.
Basic studies of this system suggest that Ang-1 is a stabilizing factor for endothelial cells; that is, it enhances endothelial cell survival and pericyte coverage. It is not truly a “proangiogenic” factor in the classic sense in that it does not promote endothelial cell proliferation. In fact, forced expression of Ang-1 in tumor cells leads to inhibited tumor growth from this “stabilizing” effect.44 In contrast, Ang-2 is a destabilizing factor that, if present with VEGF, can promote angiogenesis. Hence, Ang-2 is a rationale for inhibiting tumor angiogenesis.45 The tie-1 receptor remains an orphan receptor, with an undefined function. The Ang-1/tie-2 signaling pathway appears to be involved mainly in later stages of blood vessel formation, especially in the maturation and stabilization of vessels.46
Like VEGF and VEGF receptors, genetic disruption (“knockout”) of either Ang-1 or tie-2 leads to embryonic lethality, although both alleles of Ang-1 (or tie-2) have to be silenced (homozygous disruption), unlike VEGF.46 Studies of the role of Ang-1/tie-2 in tumor angiogenesis were hampered for many years by the inability to generate highly specific blocking antibodies or peptides. However, there are now a number of reports of the development not only of specific blocking peptides47 (“peptibodies”), which have proceeded to clinical trial assessment,45 but also monoclonal antibodies to Ang-2,48 which cause robust antitumor activity; such reagents should help considerably in clarifying the role of this system in tumor angiogenesis.
There are a number of other factors implicated in the process of angiogenesis including interleukin-8, epidermal growth factor receptor ligands, basic and acidic FGF, PDGF, among many others. However, because of the need for brevity in this chapter, we have focused on a number of factors most relevant to clinical medicine, and perhaps the future of oncology. Some of the aforementioned factors may take on increasing importance as mediators of resistance to drugs that target the VEGF pathway of angiogenesis.49,50
ENDOGENOUS INHIBITORS OF TUMOR ANGIOGENESIS
In addition to the existence of multiple molecular stimulators of angiogenesis, there are a large number of endogenous and intrinsic inhibitors of angiogenesis. The existence of such inhibitors was first surmised by Folkman3 on the basis of the observation that there are a number of tissues or organs that lack blood vessels and that also are rare sites of metastasis, such as cartilage or vitreous. It is also important to recognize the endogenous inhibitors are important in physiologic angiogenesis (wound healing, menstruation, luteal cycle) where a “stop” signal is necessary to prevent a pathologic condition.
A breakthrough in the field of endogenous inhibitors came with a series of reports by Dameron et al.51 and Bouck et al.52 beginning in 1989/1990. They reported a large glycoprotein that is a prominent member of the extracellular matrix, namely, thrombospondin-1 (TSP-1), which binds to CD36 receptors, and is a potent endogenous angiogenesis inhibitor. Moreover, the p53 suppressor gene was found to up-regulate levels of TSP-1 in various cell types, and inactivation or loss of p53 is associated with down-regulation of TSP-1 expression,51 an observation that served to link the fields of cancer genetics—specifically the role of tumor suppressor genes and oncogenes in tumor development progression—with tumor angiogenesis.3,4 Subsequently, a number of other proteins were identified as endogenous inhibitors of angiogenesis.53–58 Many, if not most, are actually proteolytically cleaved fragments of larger proteins that are members of either the clotting/coagulation cascade family (e.g., angiostatin, which is a fragment of plasminogen53) or members of the extracellular matrix family of glycoproteins. Some examples of the latter category include endostatin,54 tumstatin, and canstatin, fragments of type IV collagen.55,56 Another endogenous inhibitor is known as vasostatin, which is a fragment of calreticulin.57 Vasohibin is a secreted protein that is produced by endothelial cells on stimulation with an angiogenic stimulator such as VEGF. Vasohibin was the first example of an endogenous inhibitor that operates on the principles of a negative feedback mechanism.58 A theory that has emerged from this body of work is that tumor angiogenesis likely requires two broad functional events: the induction or elevated expression of one or more proangiogenic growth factors, such as VEGF, coinciding with the down-regulation with one or more endogenous inhibitors, such as TSP-1.3,52,59
A COOPERATIVE REGULATOR OF TUMOR ANGIOGENESIS: THE NOTCH RECEPTOR-DLL4 SIGNALING PATHWAY IN ENDOTHELIAL CELLS
During the last 5 years, the Notch/Notch ligand system has been shown to mediate embryonic and tumor angiogenesis.60–65 Notch cell surface receptors (i.e., Notch 1, 2, 3, and 4) are expressed by a number of cell types and are involved in cell fate, differentiation, and proliferation. They interact with transmembrane-bound ligands (Jagged 1, Jagged 2, DLL 1, 3, and 4) on adjacent cells. The signaling aspects are unique: on ligand binding, the intracellular domain of Notch is cleaved by gamma secretase, where the Notch intracellular domain then translocates to the nuclear and acts as a transcription cofactor. It turns out that vascular endothelial cells express Notch 1 and Notch 4 receptors and Jagged 1, DLL1, and DLL4 ligands. Of these ligands, DLL4 is the only one that is selectively expressed by endothelial cells. It is expressed in small arteries and capillaries. Gene disruption experiments have shown that Notch/DLL4 signaling is absolutely essential for vascular development and arteriogenesis in embryos. Indeed, knockout of only one DLL4 allele (haploinsufficiency) is embryonic-lethal, similar to VEGF haploinsufficiency.63 This would suggest that this system, like VEGF, would be a major stimulator of adult angiogenesis, including tumor angiogenesis. However, in some respects, Notch/DLL4 signaling is a negative regulator of tumor angiogenesis.64 Thus, it turns out that DLL4 can be significantly up-regulated in the tumor vasculature in tumors, as a consequence of VEGF function. By using a combination of neutralizing antibodies to DLL4 or other types of approaches to block DLL4 function, blood vessel formation in tumors was found to be paradoxically increased, but these vessels are largely abnormal and functionally compromised such that blood flow and perfusion are impeded and tumor hypoxia increased.60,64 This results in tumor growth inhibition. The impact of DLL4 signaling through Notch (Notch 1) is primarily restricted to tip cells at the leading edge of a growing vessel sprout or stalk, at least in the mouse retina.61
As a consequence of these biologic effects it appears that the Notch/DLL4 signaling pathway, though a “stimulator” of vasculogenesis and angiogenesis during early development, functions as an inhibitor of “productive” tumor angiogenesis in the adult. As such, it would seem that there is a sound therapeutic rationale for targeting the pathway to inhibit the growth of tumors, especially tumors that have become resistant to anti-VEGF therapies, by “converting” tumor blood vessels to a nonproductive or nonfunctional state.60–64 It would appear that angiogenesis induced by VEGF can up-regulate DLL4 in endothelial cells of newly forming blood vessels and, in so doing, act as a negative feedback mechanism to prevent excessive functional angiogenesis.62 Thus, vasohibin and DLL4 represent two negative feedback mechanisms to control/regulate tumor angiogenesis. However, the prospects of specifically targeting DLL4 received a setback recently when it was reported that chronic DLL4 blockade in mice using monoclonal antibodies caused the formation of subcutaneous vascular neoplasms and pathologic effects in organs such as the liver, thus raising critical safety concerns.65 Hence, as always, one must carefully weigh the risk-benefit ratio of agents that may exhibit toxicities due to inhibition of pathways essential to homeostatic mechanisms.
STRATEGIES FOR DEVELOPMENT OF ANTIANGIOGENIC DRUGS
Given the aforementioned information, it can be appreciated that there are a number of possible strategies that have been developed to target tumor angiogenesis. These strategies have resulted in the discovery of an unusually large and diverse number of antiangiogenics.25 The most obvious strategies would include developing drugs that neutralize proangiogenic growth factors such as VEGF, or block signaling from VEGFRs. In 1993, Kim et al.66 reported that a highly specific neutralizing monoclonal antibody to human VEGF was capable of delaying the growth of VEGF expressing transplanted human tumor xenografts in immune-deficient mice, whereas it had no antiproliferative effect on the same tumor cells in cell culture, an observation consistent with an antiangiogenic mechanism of action. This was a seminal finding that opened the field of molecularly targeted antiangiogenic drugs for the treatment of cancer.
All of the currently approved antiangiogenic drugs either target VEGF or VEGFR tyrosine kinase receptors. For example, bevacizumab, as previously discussed, is a humanized derivative of a mouse monoclonal antibody that was developed to neutralize human VEGF.26 There are also antibodies to VEGFR-2, which are in advanced clinical development,67 a murine precursors of which has been studied extensively in preclinical studies.68 Another antiangiogenic approach is the fusion of the extracellular binding domains of VEGFR-1 and -2 to an Fc backbone to create a “VEGF trap” molecule that primarily binds VEGF-A, but potentially binds other VEGFR-1 ligands as well such as PlGF.69
In addition to antibody/protein therapeutics a large number of small-molecule oral receptor tyrosine kinase inhibitors have been developed that block VEGF receptor phosphorylation. Such drugs, currently approved by the FDA, include sorafenib, sunitinib, and pazopanib.70–72 These latter drugs are also known to affect other structurally similar receptor tyrosine kinases, including PDGF-α/β, c-kit, flt-3, CSF-1R, and the serine threonine (in the case of sorafenib), raf kinase. The antibody-based drugs, which clearly target a single pathway, rarely cause tumor regressions in preclinical models, whereas in contrast, there are instances where the small-molecule multikinase inhibitors can cause regressions of even large established tumors.73 However, in such situations, the tumor responses induced may be a consequence of not only inhibition of angiogenesis, but also as a result of direct inhibition of tumor cell receptor tyrosine kinases involved in cell growth and survival. Furthermore, because they target PDGF receptors, the function of pericytes in stabilizing blood vessels may be compromised and this could conceivably also increase the initial efficacy of such drugs.74
It is important to note that at the present time it is not possible to determine which type of drug (antibody versus TKI) is optimal. With TKIs, multiple kinases may be affected, thus this class of drugs typically lead to many types of off-target toxicity and generally are more toxic than antibodies. In addition, it is more likely that there will be more patient-to-patient variation in drug exposure to small-molecule TKIs than antibodies (antibodies are dosed based on patient mass, whereas TKIs are dosed with a standard dose such as milligram per day and so forth).
A second broad approach to antiangiogenesis involves the administration of an endogenous angiogenesis inhibitor using recombinant genetically engineered protein. In this regard, there have been phase 1 clinical trials evaluating such drugs as endostatin, angiostatin, and TSP-1 peptide mimetics.75 In general, this approach has not yet shown obvious clinical benefit in clinical trials.
Another important point about antiangiogenic drugs for the treatment of cancer is the concept of “accidental” angiogenesis inhibitors.76 This refers to the idea that many anticancer drugs, both old and new, that were not developed with the intention of inhibiting angiogenesis, may in fact do so, which contributes to their overall antitumor effects. By way of example, chemotherapy drugs have been shown to have antiangiogenic effects.77 There are at least two ways this can happen: either by directly targeting dividing endothelial cells present in growing tumor blood vessels,78 or circulating bone marrow-derived endothelial progenitor cells and possibly other types of pro-angiogenic bone marrow derived circulating cells.79 The nature of these different targets is important with respect to maximizing the antiangiogenic effects of chemotherapy. For example, there is limited preclinical evidence which shows that maximum tolerated doses of a chemotherapy drug can cause apoptosis of endothelial cells in the growing tumor vasculature of transplantable mouse tumors.78 However, such a potential antiangiogenic effect is reversed during the subsequent drug-free break periods. This “repair” process may be mediated by a rapid mobilization and homing of endothelial progenitor cells to the drug-treated tumors.5,80 By shortening the break periods or even eliminating them altogether, this process can be minimized or prevented. However, this requires giving relatively low doses of chemotherapy, that is, “metronomic chemotherapy,” the antitumor effects of which can be markedly enhanced by combination with a targeted antiangiogenic drug.81,82
Metronomic chemotherapy has now been evaluated in phase 2 both randomized and nonrandomized clinical trials in a number of indications, with encouraging results, which will have to be validated in larger randomized phase 3 trials,83–85 although there are examples of successful adjuvant metronomic chemotherapylike phase 3 trials, such as the daily oral administration of UFT, a 5-fluorouracil prodrug (comprising tegafur plus uracil) for 2 years with no breaks for non–small cell lung cancer86 or breast cancer.87
ENHANCEMENT OF CHEMOTHERAPY EFFICACY AND OTHER THERAPEUTIC MODALITIES BY ANTIANGIOGENIC DRUGS
A concern in the early days of antiangiogenic drug development was that such drugs would not be useful for combination treatments involving chemotherapy or radiation therapy. By compromising blood flow/perfusion antiangiogenic drugs would “starve” tumors of oxygen, thus increasing levels of tumor hypoxia, a resistance factor to radiation and chemotherapy. However, in 1992 Teicher et al.88 reported the first of a series of studies showing that the antitumor effects of chemotherapy on transplantable mouse tumors were actually enhanced by combination with a drug known to have antiangiogenic properties. The preclinical efficacy results were subsequently confirmed in many other studies, and foreshadowed the clinical benefit successes of bevacizumab in randomized phase 3 clinical trials in which the drug was combined with various chemotherapy regimens for the treatment of metastatic colorectal,1 non–small cell lung89 and breast cancers.90 Consequently there has been considerable interest in unraveling the basis by which an antiangiogenic drug such as bevacizumab enhances the efficacy of chemotherapy. In addition, preclinical studies have shown that inclusion of an antiangiogenic drug with other therapeutic modalities—such as radiation,91 signal transduction inhibitors,92 oncolytic virus therapy,93 and vascular disrupting agents80—can enhance the antitumor activity of all these aforementioned therapies as well as others.
With respect to the mechanistic basis by which antiangiogenic drugs enhance the efficacy of other types of anticancer therapy, most studies thus far have dealt with chemotherapy or dealing with radiation therapy, and a number of hypotheses have been proposed. One proposes that a proportion of the chaotic dysfunctional tumor-associated vasculature, which is responsible for heterogeneous and often sluggish blood flow within regions of tumors, and hence regional areas of hypoxia, can be transiently “normalized” by an antiangiogenic drug, in this case, a VEGF-targeted agent.24 This can result in a paradoxical transient increase in regional blood flow, decreased hypoxia, and increased tumor cell proliferation.24 If tumors are exposed to chemotherapy or radiation during the period of “vascular normalization,” their efficacy will be increased. In addition, VEGF inhibition may decrease permeability, leading to a reduction in the high tumor interstitial fluid pressures, allowing better perfusion of tumor vessels.
The overall combined effect of all these changes would be transient episodes of increased tumor oxygenation and cell proliferation coinciding with an increased ability of the tumors to take up certain chemotherapy drugs during the “window” of vascular normalization, thus increasing the ability to affect a greater proportion of the tumor cell population than otherwise would be the case in the absence of the VEGF inhibition.24,91 However aspects of this hypothesis have yet to be confirmed clinically. Thus, paradoxical increases in blood flow may not occur in all tumors, as single-agent anti-VEGF therapy for RCC is not associated with an increase in size prior to regression. Furthermore, clinical studies involving imaging (magnetic resonance imaging, computed tomographic scan) have not demonstrated an increase in blood flow with single-agent anti-VEGF therapy.
In addition, consistent evidence that the intratumoral delivery and distribution of conventional chemotherapy drugs is improved by normalization induced by antiangiogenic drugs is still lacking, especially with respect to clinical studies. A second theory is that the presence of an antiangiogenic drug during the extended drug-free break periods following each cycle of maximum tolerated dose chemotherapy will slow down the rate of tumor cell repopulation that inevitably follows tumor shrinkage, as repopulating tumor cells would require oxygen and nutrients normally supplied by the tumor vasculature.94 In addition, there is some limited evidence that bolus injections of some cytotoxic chemotherapeutic drugs such as cyclophosphamide can cause a rapid mobilization of some of the bone marrow-derived cell populations shown in Figure 9.1. including CEPs.79 Should some of these cells then home to sites of tumor angiogenesis in the drug-treated tumors, tumor cell repopulation would be accelerated.5 There is some limited preclinical evidence that suggests this occurs after administration of a cytotoxiclike vascular disrupting agent or various chemotherapy drugs: tumor vascular disrupting agent (VDA)-induced CEPs home to the viable tumor rim that typically remains after VDA treatment and contributes to rapid tumor repopulation, a process that can be blocked by treatment with an antiangiogenic drug just before administration of the VDA.80 Such systemic CEP responses constitute a form of “rebound vasculogenesis” and can occur after maximum tolerated dose chemotherapy as well.95,96
A third theory is that tumor stem or stemlike cells (self-renewing “tumor-initiating” cells) may reside in a vascular niche within tumors and depend on the vasculature for normal function and survival, as appears to be the case for glioma stem cells.97 Disruption of the vascular niche can occur as a result of treatment with an antiangiogenic drug; the “compromised” tumor stem cell population might then be more sensitive to the chemotherapy than would otherwise be the case, provided chemotherapy retained access to the tumor.98 This possibility also highlights an emerging area of research in tumor angiogenesis, namely the link between tumor stem cells and tumor angiogenesis, given the potent tumorigenic (tumor-initiating) property of the tumor stem cell subpopulation. There is growing interest in their proangiogenic phenotype in comparison to the bulk non tumor stem cell population.97,99 On the contrary, there is also evidence in some systems that cancer stem cells may have an angiogenic-independent phenotype by adapting to hypoxic environments and using metabolic pathways that involve increased conversion of glycose to pyruvate and lactate.100 This is an area of research that merits further analysis.
A fourth theory is that chemotherapy itself might be capable of causing direct damage to the dividing, activated endothelial cells of the tumor’s growing vasculature, where the extent of such a vascular targeting effect is amplified by concurrent therapy with an antiangiogenic drug, such as an anti-VEGF antibody that would neutralize the prosurvival function of VEGF for endothelial cells, making endothelial cells more susceptible to the toxicity of chemotherapy.81,101 However, even with single-agent anti-VEGF therapy, responses are noted in patients with metastatic RCC, providing indirect evidence that single-agent anti-VEGF therapy can lead to vascular regression.102
Finally, a fifth possibility is related to the fact that VEGF may act as a direct autocrine survival factor for certain tumor cell population by virtue of their expression of receptors that can bind VEGF, for example, VEGFR-1, VEGFR-2, or neuropilin-1.103 Hence, blockade of VEGF signaling on tumor cells could conceivably directly render the cells more sensitive to chemotherapy in such cases.103,104
When considered together, these different theories serve to illustrate how difficult it is to dissect the mechanism of action of VEGF-targeted agents, despite its successes in the clinic. In addition, there are also clinical failures, and the successes observed in the clinic remain relatively modest as will be described later; thus there is a clear need to better understand mechanism(s) of action to improve efficacy and limit toxicity.
RESISTANCE TO ANTIANGIOGENIC DRUGS OR TREATMENTS
One of the theoretical advantages for antiangiogenic therapy of cancer hypothesized over 15 years ago was the possibility that this type of treatment strategy would be less susceptible to being rendered ineffective over time as a result of the development of acquired drug resistance.105 However, preclinical experiments as well as clinical outcomes with angiogenesis inhibitors have shown that acquired resistance represents a significant problem, similar in nature to virtually every other anticancer drug or treatment modality. Intrinsic resistance is also a problem. In this regard, several investigators have shown that tumor endothelial cells from tumor neovasculature may not always be genetically stable, as was initially proposed. Klagsbrun and colleagues have shown that tumor endothelial cells are aneuploid, whereas others have actually reported similar genetic mutations in tumor cells and tumor associated endothelial cells.106,107
With respect to the clinical results, with rare exception, tumors of patients that initially show good responses to VEGF-targeted agents eventually stop responding. Preclinical investigations have revealed a number of mechanisms by which resistance to a drug such as bevacizumab or VEGF-targeted TKIs can develop when such drugs are administered as monotherapies, some of which are summarized in Figure 9.3..108 Some of these resistance factors are discussed here.
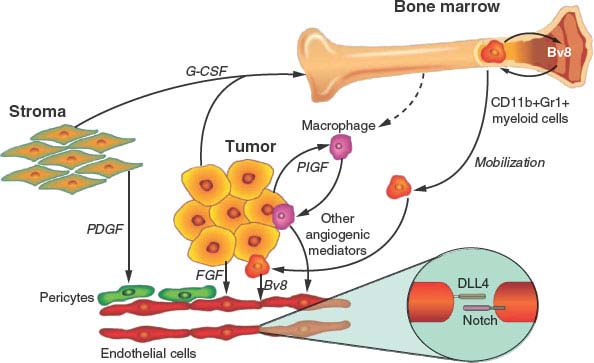
FIGURE 9.3 Some possible resistance pathways to vascular endothelial growth factor (VEGF)-targeted therapy. VEGF signaling plays a central role in tumor angiogenesis, but numerous compensatory angiogenic factors and cell types contribute to resistance to VEGF-targeted therapy. Mediators of resistance to VEGF-targeted therapy include soluble angiogenic factors such as fibroblast growth factor, placental growth factor, and Bv8; cell-bound Delta-like ligand 4 that can activate Notch on adjacent endothelial cells; pericytes that directly support endothelial cell survival; macrophages that secrete numerous angiogenic factors; and bone marrow−derived myeloid cells that also secrete soluble angiogenic factors. (From ref. 108, with permission.)
(1) Proangiogenic growth factor redundancy. There are, as summarized earlier, many different growth factors that can stimulate angiogenesis, and moreover, the number and diversity of such growth factors expressed by tumors can increase with disease progression.109 Thus, targeting a single proangiogenic pathway, especially in the context of advanced disease, by using a drug such as a monospecific antibody to VEGF or to VEGFR-2 can lead to the selection, and eventual overgrowth, of variants that can sustain angiogenesis despite persistence of VEGF/VEGFR-2 blockade.50,110 By way of example, an alternative proangiogenic growth factor, such as bFGF, can assume control and begin to induce tumor angiogenesis during anti-VEGFR-2 antibody therapy, even though decreases in phosphorylated VEGFR-2 are detected in tumors that initially responded to the drug.50,110 The bFGF was induced in the tumor cell population, probably as a consequence of elevated levels of hypoxia induced by drug treatment, and thus up-regulation of various growth factors known to be regulated by hypoxia.50 Such findings would appear to support a theoretical advantage of using multitargeting TKI antiangiogenic drugs that block several proangiogenic pathways simultaneously as a means of significantly delaying or circumventing this type of acquired resistance. However, as previously mentioned, resistance to such drugs (e.g., sunitinib or sorafenib, as in RCC) eventually occurs in all patients who initially respond to treatment. One recent report implicated up-regulation of interleukin-8 as a mechanism for resistance to sunitinib in a model of RCC.111 In some cases the source of a compensatory growth factor may be the tumor stroma, such as fibroblast-derived PDGF-C.110 In addition, drugs such as sunitinib can induce elevated levels of multiple cytokines, chemokines, and growth factors such as granulocyte-colony stimulating factor, stromal-derived factor-1-1, PlGF, and VEGF in a tumor-independent fashion, and it is conceivable, but not yet proven, that these drug-induced changes could contribute to acquired resistance.112
(2) Selection for hypoxia-resistant cells. Cancer cells, as a result of certain genetic mutations (e.g., p53 mutation/inactivation), can acquire an enhanced ability to survive under relatively hypoxic conditions, as would be expected to occur during an effective and long-term antiangiogenic therapy.113 Thus, over time there could be a selection for mutant/variant subpopulations that depend less on tumor angiogenesis for survival, and possibly even cell growth.113
(3) Co-option of normal organ vasculature. It has been proposed that the ability of tumors to grow in certain vascular-rich organs such as the lung, brain, or liver might not be affected significantly by antiangiogenic drugs by virtue of the tumor cells exploiting (“co-opting”) the existing mature normal vasculature to obtain the necessary oxygen and nutrients for robust growth11; this might also contribute to “mixed” responses in patients in whom tumors in one organ location respond to antiangiogenic treatment, but do not do so in a different organ.
(4) Vascular remodeling. Antiangiogenic drugs tend to preferentially target relatively immature growing neovasculature and have much reduced or even no efficacy on established/more mature vessels.114 It has been reported that antiangiogenic therapy in preclinical tumor models can accelerate the maturation and remodeling of blood vessels, which become progressively less sensitive to the therapy.114 The remodeled vasculature may be driven by increased expression of various factors that contribute to vessel stabilization and maturation (e.g., PDGF-BB and angiopoietin-1).114
With respect to development of new strategies that could have promise in dealing with tumors that are either intrinsically resistant to VEGF-targeted therapies, several strategies are being evaluated, such as sequential or salvage therapy with different antiangiogenic drugs (e.g., patients whose RCC stops responding to sunitinib may respond to sorafenib, or vice versa).70 Combining VEGFR-2 pathway targeting drugs with other antiangiogenic drugs that block a different, complementary pathway (e.g., the tie-2/Ang2 pathway),115 may be a promising strategy. Similarly, one might use drugs that target HIF-1α and the hypoxic tumor microenvironment, a consequence of VEGF-targeted therapies.
BIOMARKERS FOR TUMOR ANGIOGENESIS AND ANTIANGIOGENIC THERAPY
A challenge associated with the development and clinical use of antiangiogenic drugs, which is similar in nature to many other types of anticancer therapeutic modalities, especially “targeted” therapies, is the need for predictive and surrogate biomarkers to improve overall therapeutic benefit, including increasing efficacy, reducing toxicity, and improving cost-effectiveness.116 It is important to make the distinction between predictive and surrogate markers. Predictive markers are identified prior to treatment to identify patients who may or may not benefit from therapy (predictive markers may also be used to identify patients who may develop toxicity). A surrogate marker is one that changes after initiation of therapy whereby the change may indicate target modulation and, hopefully, clinical benefit.116
Although there are a number of in vivo assays to monitor angiogenesis and hence inhibition of angiogenesis that are commonly used in mice, none of these are of practical use for use in humans.
Potential biomarkers for antiangiogenic therapies markers under investigation include circulating proteins, for example, VEGF or other proangiogenic growth factors, and soluble VEGF receptors.116–118 In addition, circulating cells thought to be relevant to angiogenesis have been studied, including circulating endothelial cells and circulating endothelial progenitor cells.4,9 Finally, another intensively studied approach is based on noninvasive imaging of blood flow or vascular permeability using such methods as dynamic contrast enhanced magnetic resonance imaging, computed tomographic scans incorporating flow parameters, or high-frequency microultrasound, among others.103,116 Thus far, none of these approaches has yet been validated in prospective randomized clinical trials. However, recent studies have implicated the possibility that certain single nucleotide polymorphisms in angiogenesis-related genes (e.g., the vegf gene) may have promise as predictive markers for VEGF pathway targeting drugs such as bevacizumab, both for predicting clinical benefit as well as certain toxicities such as hypertension, a common side effect of VEGF inhibition.119,120 Indeed, elevated hypertension is also currently being evaluated as a relatively simple and inexpensive surrogate biomarker, although this remains a point of controversy.121
To illustrate the nature of the considerable challenges involved in developing biomarkers for antiangiogenic drugs, attempts to exploit VEGF as a predictive marker for possible clinical benefit in patients receiving anti-VEGF monoclonal antibody (bevacizumab) therapy provides a compelling example. There is abundant literature reporting elevated levels of tumor VEGF are associated with a poor prognosis, so it might be anticipated that examining VEGF levels in tumors or in the circulation would be a relatively single predictive assay: the higher the levels of VEGF, the target of bevacizumab, the more likely a patient would benefit from bevacizumab therapy. However, neither VEGF expression in tumor tissue or circulating VEGF levels is predictive of clinical benefit for patients receiving either bevacizumab or other drugs that target the VEGF pathway.122,123
ANTIANGIOGENIC/ ANTI-VEGF DRUG-BASED CLINICAL TRIALS
Research over the last two decades has shed a tremendous amount of light on the process of angiogenesis, which in turn has led the successful application of this knowledge to the care of patients with advanced-stage malignancies. All of the FDA-approved drugs considered to be antiangiogenic interfere with VEGF signaling, whereas the effectiveness of other agents remains to be determined. Because VEGF plays such diverse roles in regulating vascular development, function, and morphology, it is important at this stage in time to differentiate anti-VEGF therapy from other agents considered to be antiangiogenic. Anti-VEGF therapy can theoretically be of benefit to patients by numerous mechanisms,124 as discussed earlier in this chapter. Thus, it is appropriate to refer to agents that inhibit VEGF signaling as “anti-VEGF agents” or “VEGF-targeted therapies,” providing a distinction from “generic” antiangiogenic agents that primarily target tumor endothelial cell proliferation.
Anti-VEGF therapy, despite all its high profile, does not always lead to patient benefit. A few principles derived from phase 3 clinical trial results deserve mention (Tables 9.1 and 9.2), with details of clinical trials being presented in other chapters in this text. First, for tumors other than RCC, anti-VEGF therapy is of benefit only to patients when combined with chemotherapy. The benefit to patients with RCC may be because there is a well-defined, dominant molecular alteration (loss of von Hippel–Lindau function) leading to tumors highly dependent on VEGF signaling. Second, despite the fact that anti-VEGF therapy appears to augment the effects of chemotherapy, this is not always the case. Tyrosine kinase inhibitors for patients with various types of metastatic cancer have so far failed to demonstrate improved efficacy over chemotherapy alone in multiple randomized phase 3 clinical trials, many of which have not yet been reported or published.125,126 In addition, in trials in patients with metastatic breast cancer, bevacizumab augments the effects of paclitaxel in patients in the front-line setting, but in later lines of therapy the addition of bevacizumab provided no benefit when added to capecitabine. Lastly, anti-VEGF therapy leads to specific and sometimes unexpected toxicities, such as hypertension, proteinuria, bowel perforations, hemorrhage, arteriothrombotic events, and others. Some of these adverse effects may be due to our understanding of basic biology. For example, many investigators believe that hypertension associated with anti-VEGF therapy is due to inhibition of endothelial cell-derived nitric oxide, known pathway mediated by VEGFR-2 activation. However, the basis of other toxicities, such as bowel perforation, remain a mystery.127
TABLE 9.1
PHASE 3 TRIALS: CHEMOTHERAPY WITH OR WITHOUT VASCULAR ENDOTHELIAL GROWTH FACTOR (VEGF) TARGETED THERAPIES
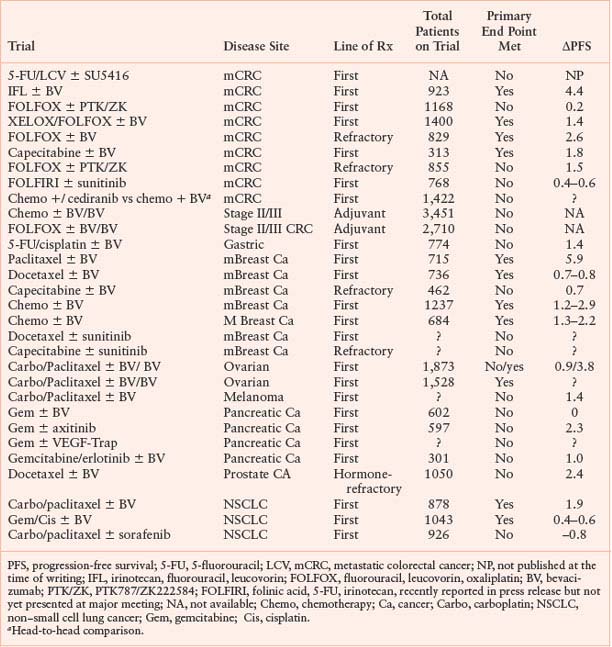
TABLE 9.2
PHASE 3 TRIALS: VASCULAR ENDOTHELIAL GROWTH FACTOR-TARGETED THERAPIES (SINGLE AGENT OR WITH A BIOLOGIC)
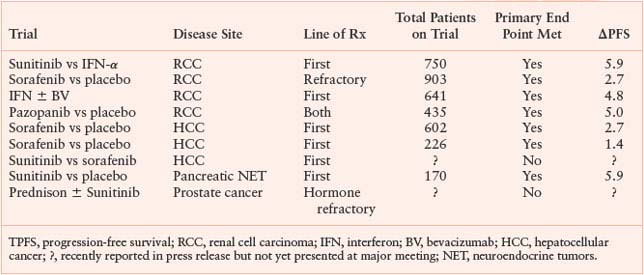
It is important to point out that the benefits obtained with anti-VEGF/angiogenic therapy are incremental; cures are rare and tumor dormancy, if it occurs, is short-lived and rarely lasts beyond a year. The use of anti-VEGF therapy as “maintenance therapy” (after maximal tumor response with chemotherapy) has only recently been tested in a clinical trial.128 Furthermore, the use of anti-VEGF/angiogenic therapy in the adjuvant setting is under study, but with one exception (Table 9.1), results from most such trials will not be available for several years. The exception comes from the results of the first randomized phase 3 trial involving an antiangiogenic drug, which was recently announced.129 The trial evaluated bevacizumab in combination with chemotherapy for 6 months followed by bevacizumab maintenance therapy for 6 additional months. The trial did not meet its primary end point of a progression-free survival benefit at 3 years despite prior interim analyses showing a transient benefit in the bevacizumab arm.129 This benefit gradually disappeared over time, suggesting that there may be a change in the biological aggressiveness of tumor growth after the bevacizumab therapy has been completed.
Similarly, many recent phase 3 trials of bevacizumab plus chemotherapy have shown a benefit in progression-free survival but not overall survival. This has been reported for a number of trials in metastatic breast cancer, for example,90 and also for trials in colorectal cancer,130 gastric cancer,131 and ovarian cancer.128 The lack of overall survival benefit may be because of subsequent lines of therapy diluting the effect of front-line therapy. However, others have hypothesized that these findings suggest a possible change in the biology of tumor growth after an initial tumor response that reduces some of the initial clinical benefit. In this regard several recent preclinical studies have shown that there may be circumstances in which treatment with an antiangiogenic drug may accelerate tumor growth once treatment is stopped, and may also cause an increase in tumor cell invasion and/or metastasis.112,132 With the possible exception of glioblastoma,133,134 evidence for such increased malignant aggressiveness in the clinic has not yet been reported when drugs such as bevacizumab (plus chemotherapy) or single-agent antiangiogenic TKIs are used to treat various types of cancer. Moreover, if and when such increases in malignant aggressiveness occur, this does not mean that survival is decreased, but rather that the overall clinical benefits attained may be less than would otherwise be the case.132 One theory to explain increases in malignant aggressiveness is that the elevated tumor hypoxia induced by antiangiogenic drug treatments will result in up-regulation of HIF-1α, which regulates numerous genes that contribute to tumor cell motility, invasion, and metastasis.132,135,136
LOOKING AHEAD: NEW TARGETS, NEW DRUGS, AND NEW STRATEGIES FOR ANTIANGIOGENIC THERAPY
We have stressed that, thus far, all approved antiangiogenic drugs involve the VEGF pathway as the only or primary target. Despite their successes, the limitations of such drugs highlight the need to develop alternative or complementary approaches to blocking tumor angiogenesis and improving the effects of existing drugs. With respect to new targets, there is currently considerable effort being put into defining molecular signatures of activated endothelial cells that may reveal new drivers of angiogenesis and/or promising molecular targets, especially those that are independent of the VEGF pathway. Some promising developments in this regard include the discovery and functional characterization of microRNAs—small noncoding RNA molecules that regulate gene expression at the posttranscriptional level—in vascular endothelial cells or tumor cells, and which thus can either stimulate or suppress angiogenesis.137–139 For example, members of the microRNA-17-92 cluster have an antiangiogenic effect in endothelial cells140 whereas mir-126, an endothelial-specific microRNA, can contribute to activation of VEGF signaling.141
Likewise, genomic and proteomic profiling of endothelial cells,142 especially those isolated from the tumor vasculature, represents another approach being undertaken to uncover new molecular mediators of tumor angiogenesis.15 With respect to new strategies, numerous possibilities exist, among them is the combining of antiangiogenic drugs with therapeutic modalities other than chemotherapy. For example, VEGF may have an impact on regulating components of the immune system, acting mainly as an immunosuppressive-regulating element.143 Hence, VEGF inhibition may stimulate the immune system, making VEGF pathway targeting drugs ideal to combine with tumor vaccines or other immunotherapeutic methodologies. Many other types of combination treatment involving antiangiogenic drugs are currently under preclinical and clinical investigation.93 Lastly, identifying patients likely to respond (or not respond) to current antiangiogenic therapies will lead to improvements in outcomes in those patients deemed likely to benefit from such therapies, while allowing other patients to be eligible for alternative antineoplastic approaches.
Selected References
The full list of references for this chapter appears in the online version.
1. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350: 2335.
2. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971;285:1182.
3. Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov 2007;6:273.
4. Kerbel RS. Tumor angiogenesis. New Engl J Med 2008; 358:2039.
5. Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science 2006;312:1171.
6. Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol 2006;3:24.
14. St. Croix B, Rago C, Velculescu V, et al. Genes expressed in human tumor endothelium. Science 2000;289:1197.
22. Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest 2003;111:1287.
23. Xian X, Hakansson J, Stahlberg A, et al. Pericytes limit tumor cell metastasis. J Clin Invest 2006;116:642.
24. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005;307:58.
25. Kerbel RS, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2002;2:727.
26. Ferrara N. Timeline: VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer 2002;2:795.
27. Sawamiphak S, Seidel S, Essmann CL, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature 2010;465:487.
28. Senger DR, Galli S, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983;219:983.
29. Leung DW, Cachianes G, Kuang W-J, Goeddel DV, Ferrara N. Vascular Endothelial Growth Factor is a secreted angiogenic molecule. Science 1989;246:1306.
32. Ellis LM. The role of neuropilins in cancer. Mol Cancer Ther 2006;5:1099.
34. Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res 2006;312:549.
36. Ferrara N, Carver-Moore K, Chen H, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996;380:439.
37. Carmeliet P, Ferreira V, Breier G, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996;380:435.
38. Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995;1(10): 1024.
41. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003;3:721.
46. Hanahan D. Signaling vascular morphogenesis and maintenance. Science 1997;277:48.
47. Oliner J, Min H, Leal J, et al. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell 2004;6:507.
48. Brown JL, Cao ZA, Pinzon-Ortiz M, et al. A human monoclonal anti-ANG2 antibody leads to broad antitumor activity in combination with VEGF inhibitors and chemotherapy agents in preclinical models. Mol Cancer Ther 2010;9:145.
49. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008;8:592.
50. Casanovas O, Hicklin D, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late stage pancreatic islet tumors. Cancer Cell 2005;8:299.
52. Bouck N, Stellmach V, Hsu SC. How tumors become angiogenic. Adv Cancer Res 1996;69:135.
60. Noguera-Troise I, Daly C, Papadopoulos NJ, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006;444:1032.
61. Hellstrom M, Phng LK, Hofmann JJ, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007;445:776.
66. Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993;362:841.
68. Prewett M, Huber J, Li Y, et al. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res 1999;59:5209.
69. Lockhart AC, Rothenberg ML, Dupont J, et al. Phase I study of intravenous vascular endothelial growth factor trap, aflibercept, in patients with advanced solid tumors. J Clin Oncol 2010;28:207.
70. Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol 2009;10:992.
78. Browder T, Butterfield CE, Kraling BM, Marshall B, O’Reilly MS, Folkman J. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res 2000;60:1878.
80. Shaked Y, Ciarrocchi A, Franco M, et al. Therapy-induced acute recruitment of circulating endothelial progenitor cells to tumors. Science 2006;313:1785.
81. Klement G, Baruchel S, Rak J, et al. Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest 2000;105:R15.
89. Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 2006;355:2542.
90. Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 2007;357:2666.
97. Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007;11: 69.
103. Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 2005;23:1011.
109. Relf M, LeJeune S, Scott PA, et al. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res 1997;57:963.
110. Ferrara N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev 2010;21:21.
112. Ebos JML, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009;15:232.
116. Murukesh N, Dive C, Jayson GC. Biomarkers of angiogenesis and their role in the development of VEGF inhibitors. Br J Cancer 2010;102:8.
120. Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol 2009;6:465.
132. Paez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009;15:220.
134. Norden AD, Young GS, Setayesh K, et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology 2008;70:779.
136. Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003;3:347.
137. Heusschen R, van GM, Griffioen AW, Thijssen VL. MicroRNAs in the tumor endothelium: novel controls on the angioregulatory switchboard. Biochim Biophys Acta 2010;1805:87.
139. Bonauer A, Boon RA, Dimmeler S. Vascular microRNAs. Curr Drug Targets 2010;11:943.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


