Edward N. Janoff, Daniel M. Musher Keywords antimicrobial resistance; bacteremia; bronchitis; capsular polysaccharides; colonization; common variable immunodeficiency; conjugate vaccines; gram-positive coccus; IgA1 protease; immunodeficiency; meningitis; mucosal immunity; neutropenia; opportunistic infection; otitis media; pneumococcal pneumonia; Pneumococcus; pneumolysin; pneumonia; postsplenectomy sepsis; serotypes; sinusitis; Streptococcus pneumoniae; vaccines
Streptococcus pneumoniae
Long recognized for causing asymptomatic colonization and as a prominent cause of pneumonia, bacteremia, meningitis, sinusitis, and otitis media, Streptococcus pneumoniae is likely the most common worldwide cause of serious bacterial respiratory infection in both children and adults. Antimicrobials and vaccines have substantially reduced the incidence of, and morbid outcomes from, pneumococcal infection, but acquisition of antibiotic resistance and the more limited impact of vaccines on mucosal disease, including pneumonia and otitis media, as well as the emergence of nonvaccine serotypes, provide challenges for ongoing control of this prevalent and invasive pathogen.
History
S. pneumoniae has played a prominent role in the history of microbiology. Identified concurrently in 1881 in France by Pasteur and in the United States by Sternberg, this bacterium was soon recognized as the most common cause of lobar pneumonia and became known as the pneumococcus. Based on its appearance in Gram-stained sputum, the name Diplococcus pneumoniae was assigned in 1926 and finally Streptococcus pneumoniae in 1974, based on its morphology during growth in liquid medium.
S. pneumoniae was the first organism to be recognized as showing characteristics of a prototypic extracellular bacterial pathogen by replicating extracellularly in mammalian tissues. Most of the resistance to uptake by phagocytic cells is related to the polysaccharide capsule. In the early 1890s, Felix and Georg Klemperer showed that immunization with killed pneumococci protected rabbits against subsequent pneumococcal challenge and that protection could be transferred by infusing serum (“humoral” substance) from immunized rabbits into naïve recipients. Subsequently, sera from persons recovered from pneumococcal pneumonia conferred the same degree of protection. The basis for this immunity was shown by Neufeld and Rimpau to be the presence of factor(s) in serum that facilitate ingestion by white blood cells (WBCs), a process they called opsonization, derived from the Greek word for preparing food.1 These observations provided the basis for what we now call humoral immunity. Individual capsular serotypes were recognized when injection of killed organisms into a rabbit stimulated the production of serum antibody that agglutinated and caused capsular swelling (“Quellung”) of the immunizing strain, as well as some, but not all, other pneumococcal isolates. Early in the 20th century, serotypes 1, 2, and 3 were distinguished, and all other pneumococci were called group 4.
In the first decade of the 20th century, the concept of humoral immunity was used to address the problem of epidemic lobar pneumonia that each year affected as many as 1 in 10 African miners.1,2 A program of immunization with whole killed pneumococci substantially reduced the incidence of pneumonia. In the 1920s, Heidelberger and Avery3 demonstrated that the protective antibody reacted with surface capsular polysaccharides, the first nonprotein antigen identified. Felton4 prepared the first purified pneumococcal capsular polysaccharides (PPSs) for immunization of human subjects, and mass vaccination with Felton’s type 1 polysaccharide aborted an epidemic of pneumonia at a state hospital in Massachusetts in the winter of 1937 to 1938.5 Thus, specific bacterial polysaccharide antigens could be used to stimulate antibodies that conferred protection against epidemic human infection. These observations were confirmed during World War II when MacLeod and co-workers6 showed that vaccinating military recruits with capsular material from four serotypes of S. pneumoniae greatly reduced the incidence of pneumonia caused by serotypes in the vaccine but not by other serotypes.
S. pneumoniae also played a central role in the discovery of DNA. Experiments by Griffith7 in the 1920s revealed that intraperitoneal injection into mice of live, unencapsulated (and, therefore, avirulent) pneumococci, together with heat-killed encapsulated pneumococci, led to the emergence of encapsulated virulent bacteria, a process he called transformation. This observation remained unexplained until the 1940s, when Avery and co-workers8 provided conclusive evidence that these mutants had recovered the capacity to produce capsule by taking up nucleic acids from killed, virulent organisms. Therefore, this discovery was the first to identify a functional transferable material (DNA) that encoded for phenotype.
Finally, pneumococcal infections were among the first to be treated with an antimicrobial agent, in this case optochin (ethylhydrocupreine), a quinine derivative. The organism was also among the first to develop resistance to such therapy, resulting in failure of treatment both in experimental animals and in humans. These findings were premonitory for the remarkable resilience of bacteria, and S. pneumoniae in particular, that have evolved and adapted over many millions of years to subvert our efforts to control and eradicate them with antimicrobials and vaccines developed in the last 100 years.
Microbiology
S. pneumoniae is a gram-positive coccus that replicates in chains in liquid medium but appears as lancet-shaped diplococci in clinical specimens. The organism is catalase negative, but generates hydrogen peroxide (H2O2) via a flavoenzyme system and therefore grows better in the presence of a source of catalase, such as red blood cells. Pneumococci produce pneumolysin (formerly called α-hemolysin), which breaks down hemoglobin into a green pigment that surrounds the colonies during growth on blood and chocolate agar plates, a phenomenon still termed α-hemolysis. Pneumococci may be identified in the microbiology laboratory by three reactions: (1) α-hemolysis of blood agar, (2) susceptibility to optochin, and (3) solubility in bile salts (sodium deoxycholate). Some pneumococci are optochin resistant, and a newly recognized species, Streptococcus pseudopneumoniae, which is associated with exacerbation of chronic obstructive pulmonary disease or pneumonia, is optochin susceptible during growth in room air at 37° C but optochin resistant when grown in the presence of increased carbon dioxide. These factors have led to greater reliance on the use of bile solubility and reliable commercial DNA probes for the ribosomal RNA (rRNA) gene for definitive identification.
Peptidoglycan and teichoic acid are the principal constituents of the pneumococcal cell wall9 (Fig. 201-1). Peptidoglycan consists of long chains of alternating N-acetyl-d-glucosamine and N-acetylmuramic acid, from which extend chains of four to six amino acids called stem peptides. Stem peptides are cross-linked by pentaglycine bridges, which provide substantial strength to the cell wall. Teichoic acid, a carbohydrate polymer that contains phosphorylcholine, is covalently linked to the peptidoglycan on the outer surface of the bacterial wall and protrudes into the capsule. Teichoic acid and tightly adherent fragments of peptidoglycan make up C-polysaccharide, a substance present in all pneumococci but only in a few species of viridans streptococci. C-polysaccharide reacts with acute-phase reactants in the blood during inflammation, including C-reactive proteins. Many proteins are expressed on the pneumococcal cell surface. Of particular importance in the pathogenesis of pneumococcal disease are those that bind to choline, including surface proteins A and C (PspA, PspC), surface adhesin (choline-binding protein) A (CbpA), choline-binding protein C, and proteins involved in receptivity to DNA acquisition or competence (Table 201-1).
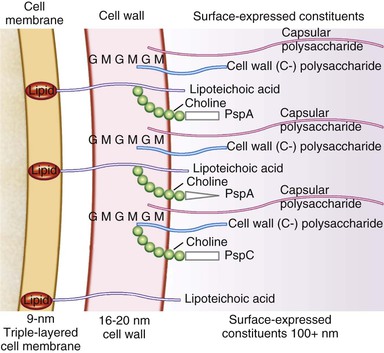
TABLE 201-1
Role of Pneumococcal Constituents as Virulence Factors*
| PNEUMOCOCCAL CONSTITUENT | MECHANISM | STRENGTH OF EVIDENCE AS A VIRULENCE FACTOR | |
| Antibody Prevents Disease† | Mutants Lack Virulence | ||
| Capsular polysaccharide | Prevents phagocytosis; activates complement | 4+ | 4+ |
| Cell wall polysaccharide | Stimulates inflammation by strongly activating complement and stimulating release of cytokines | 0 | ND |
| Pneumolysin | Cytotoxic; activates complement, cytokines | 2-3+ | 2-3+ |
| PspA | Inhibits phagocytosis by blocking activation and deposition of complement on bacterial surface | 2+ | 2+ |
| PspC | Inhibits phagocytosis by binding complement factor H | 1-2+ | 1-2+ |
| PsaA | Mediates adherence | 1-2+ | 1-2+ |
| Autolysin | Causes bacterial disintegration, releases components | 1+ | 2+ |
| Neuraminidase | Possibly supports adherence | 0-1+ | 0-1+ |

* The grading system is subjective and indicates (on a scale of 1+ to 4+) the stringency and importance of the demonstrated effect. For discussion and references, see the text.
† Animal models only, except capsular polysaccharides.
ND, not done; Psa, pneumococcal surface adhesin; Psp, pneumococcal surface protein.
Nearly every clinical isolate of S. pneumoniae contains an external polysaccharide capsule; unencapsulated isolates have mainly been implicated in outbreaks of conjunctivitis.10 Capsules11 (see Fig. 201-1) are made of repeating oligosaccharides that are synthesized within the cytoplasm, polymerized, and transported to the bacterial surface by cell membrane transferases. These polysaccharides are covalently bound to peptidoglycan and C-polysaccharide, which explains the difficulty of separating capsular from cell wall polysaccharide in vaccine preparations. Genetic control of this complex set of events has been elucidated for some serotypes; for example, a cassette of 15 genes that function as a single transcriptional unit is responsible for encapsulation in serogroup 19.12 More than 92 serotypes of S. pneumoniae have been identified on the basis of antigenic differences in their capsular polysaccharides. Among the multiple genes that encode production of individual capsules, some are specific for individual polysaccharides, whereas others are conserved among nearly all pneumococci and even some other streptococci.13
Immunization of rabbits with a pneumococcus of a specific capsular type stimulates the appearance of antibodies that cause agglutination and create a hydrophobic border around the capsule. This latter reaction, called the Quellung reaction, renders the capsule refractile and therefore more readily visible under the microscope. Because serum antibody is the basis for identifying these types of pneumococcus, they are called serotypes. In the American numbering system, serotypes are numbered sequentially in the order in which they were identified historically. The more widely accepted Danish numbering system distinguishes 46 serogroups, with groups containing antigenically related serotypes. For example, Danish serogroup 19 includes serotypes 19F, 19A, 19B, and 19C (the letter F indicates the first member of the group to be identified, followed by A, B, C, etc.), which in the American system would be serotypes 19, 57, 58, and 59, respectively. The serotypes that most frequently caused human disease were the earliest to be identified and the first to be assigned numbers, which explains why the lower-numbered serotypes are generally more likely to be implicated in human infection. Serotyping was clinically relevant in the preantibiotic 1920s to 1930s, when capsule-specific horse antisera were administered for therapy. Today, serotypes are of great interest from epidemiologic and public health standpoints, especially as targets for current and newer vaccines, and for understanding pathogenesis, but they have less relevance for the clinician in an individual case of pneumococcal infection.
An important property of S. pneumoniae is its capacity, as part of its quorum-sensing mechanism, to express a competence-sensing protein and internalize DNA from other pneumococci or from other bacterial species.14 This horizontal transfer of genetic information to pneumococci, called transformation, enables pneumococci to acquire new traits. Of note, a pneumococcus of one serotype can acquire DNA that encodes a different capsular polysaccharide, thereby changing its serotype. This exchange of genetic information occurs under experimental conditions and, more importantly, also in nature.15 Thus, in addition to a highly conserved genetic core, S. pneumoniae supports a large number of noncore genes that provide remarkable diversity of genetic loci between isolates, particularly related to antimicrobial targets and targets of immune recognition (capsular polysaccharides and adhesins).16,17
Epidemiology
The spectrum of pneumococcal infections can range from asymptomatic colonization to mucosal disease (otitis media, sinusitis, pneumonia) to invasive infections (infection of previously sterile sites). Although otitis media may be the most common clinical manifestation, pneumococcal pneumonia has the greatest impact on morbidity and mortality. Indeed, pneumonia is the leading cause of death of children worldwide, accounting for 1 in 5 deaths, and S. pneumoniae is the leading cause of bacterial childhood pneumonia. Pneumococcal disease (pneumonia, sepsis, and meningitis) is the leading cause of bacterial deaths in young children, causing up to one million deaths per year (more than acquired immunodeficiency syndrome [AIDS], malaria, and measles combined) in those younger than 5 years,18 particularly in resource-limited countries,. After a nadir at 5 to 17 years, the incidence of pneumococcal pneumonia increases progressively with age. In adults in Kenya, S. pneumoniae is identified in one half of cases of lobar and multilobar pneumonia19 and is the leading identified cause of bacterial pneumonia in older adults in industrialized countries.
Invasive pneumococcal infections are most prominent at the extremes of life (Fig. 201-2).20 Consistent results in multiple ethnic and geographic groups highlight the tremendous impact of age on the incidence of bacteremia.1,21,22 In the preconjugate vaccine era (before 2000), pneumococcal bacteremia occurred at an approximately 10-fold higher rate among children younger than 2 years than among adults in the general population and in all populations studied, independent of ethnicity (e.g., White Mountain Apaches) or underlying disease (sickle cell disease, splenectomy, human immunodeficiency virus [HIV] infection).23 These results are likely derived in part from low levels of capsule-specific antibody because children younger than 2 years initially show a limited ability to recognize and generate protective antibodies to PPSs. Infants in the first few months of life tend to be relatively spared. This early partial protection is consistent with the passive transfer of capsule-specific mucosal immunoglobulin A [IgA] and innate factors from breast milk to the upper respiratory tract and specific IgG to serum transplacentally through cord blood, levels of which decline by 6 months of age. Implementation of early pneumococcal vaccination at 2, 4, and 6 months of age for infants in the United States has already had a major impact on the rate of disease among the very young.
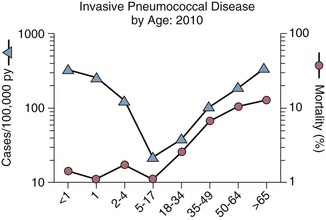
Among children 6 months to 2 years of age, invasive pneumococcal disease is diagnosed primarily when blood cultures are obtained to evaluate for fever. Many of the affected children have no apparent focus of infection and are not hospitalized. This syndrome is called “primary bacteremia” and is associated with a poor outcome if proper antibiotic therapy is not given. Hospitalization is more common with associated underlying cardiac, respiratory, and neurologic disease. Unlike in adults, among whom bacteremia is most often a complication of pneumonia (>80%), bacteremic pneumonia in young children accounts for 28% to 77% of pneumococcal bacteremias in developing countries and 13% to 60% in more industrialized countries.24 Primary bacteremia accounts for 61% to 70% of invasive pneumococcal disease in infants in the United States but is uncommonly diagnosed in developing countries; associated mortality in the United States is typically low (<1%) but varies, and a third of cases resolve spontaneously. These frequencies in children have declined by approximately 90% in the United States after the polysaccharide-protein conjugate vaccine was introduced. Whether the relatively low levels of serum antibodies to PPSs, to other protein antigens, or innate factors in healthy adults underlie their relatively low incidence of pneumococcal disease is not well understood.25,26
Adults older than 65 years comprise about 15% of the population but experience one third of all cases of invasive pneumococcal disease (≈15,000 episodes/yr, with ≥15% mortality). Most invasive cases result from complications of pneumonia (70% to >80%), but 5 to 10 times as many older adults experience pneumococcal pneumonia without bacteremia. Thus, the mortality associated with S. pneumoniae is likely much greater in the population than the numbers for invasive disease predict.27 Mortality also increases substantially with age (see Fig. 201-2) and is more than two- to fivefold greater among adults with underlying disease (advanced lung, heart, kidney, liver disease, diabetes, asplenia, solid and hematologic malignancies, and immunosuppression) than in healthier older adults.28
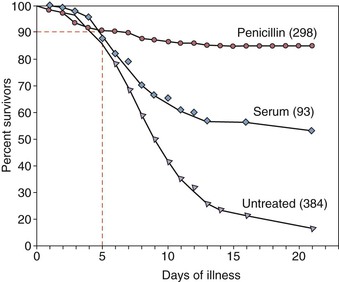
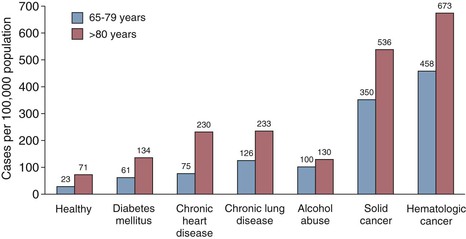
Deaths from pneumococcal bacteremia tend to occur quickly, often in the first day to week27,29,30 of hospitalization. Despite advances in antimicrobial therapy, the 5% to 10% early mortality with pneumococcal bacteremia has remained constant over the last century (Fig. 201-3). Although chronologic age itself is a factor, most pneumococcal disease in older adults and mortality occur in subjects with diabetes; underlying organ dysfunction (e.g., liver, kidney, heart, lung); specific immunologic diseases, such as B-cell cancers (chronic lymphocytic leukemia, multiple myeloma); and use of immunosuppressive medication (Fig. 201-4).31–33 These data suggest an independent or additive contribution of underlying disease to the increased risk of serious infection with age.
Pneumococcal disease is not generally regarded as highly contagious. Pneumococci are transmitted from one person to another as a result of close contact,34 such as among toddlers in daycare centers,35,36 but many steps intervene between spread of organisms, colonization, and development of disease.34 However, community-wide epidemics are well recognized in the meningitis belt in West Africa, where capsular serotype 1 predominates,37 and smaller outbreaks of disease, often caused by capsular serotypes 1, 5, and 12F,38,39 are well documented among adults in crowded living conditions, such as in military camps,40 prisons,41 shelters for the homeless,42 and nursing homes.43 In contrast, close contact in schools or the workplace is generally not associated with outbreaks of pneumococcal infection.34,44 A very high incidence of invasive infections occurs at all ages among African Americans,45 Native Americans,46,47 and Alaskan48 and Australian aboriginals,49 presumably related to both socioeconomic and genetic factors. Infants from these populations and other disadvantaged members of developed societies50 are more likely to be colonized and with high numbers of pneumococci, even within the first few weeks of life, adding exposure to their risk for disease.
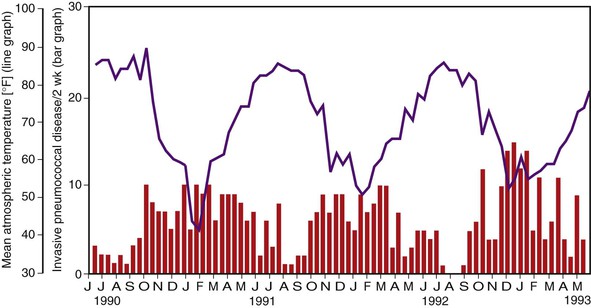
Pneumococcal colonization and disease occur with a seasonal pattern, with a midwinter increase, although pneumococci can be recovered from healthy children and adults throughout the year. The incidence of pneumococcal otitis media clusters largely from November to April.51,52 Bacteremia53,54 shows a clear midwinter peak in temperate climates (Fig. 201-5). The association may relate in part to the colder temperatures and lower humidity, which appear to predispose to transmission of respiratory viruses, such as influenza,53–55 and perhaps closer contact indoors. However, in Houston, Texas, invasive disease in children coincides with the school year, from September through May, sparing the summer months and with no clear midwinter peak.54 The seasonal distribution is less prominent in tropical climates.
Pathogenetic Mechanisms
Colonization
The prevalence of pneumococcal colonization attests to the success of this organism in adapting to adherence and survival in the nasopharynx. The vast majority of serial colonizing episodes remain asymptomatic, but most symptomatic infections are likely initiated after asymptomatic colonization.52,56 Nasopharyngeal colonization with pneumococci begins in the first weeks of life. The prevalence of colonization increases from less than 10% to a peak at 70% to 100% at age 1 year, persists through the second and third year of life, and decreases thereafter to adult rates of approximately 10%. Defining such rates depends on the sampling location, methods, and culture,57 including more recent use of high-throughput sequencing of the 16S rRNA gene from mixed mucosal samples.58 Duration of carriage can range from 1 week to 6 months. In adults, an individual serotype persists for shorter periods, usually 2 to 4 weeks,59 but sometimes for several months.1
Living in resource-limited countries and the presence of siblings in the home are consistent risks for colonization.50,60 Crowding (e.g., in home, daycare, barracks),61 lower socioeconomic status, and ethnic background, as mentioned earlier, have been associated with high carriage, as have smoke exposure (passive or active smoking41 and cooking fires in the home), antibiotic use (with associated risk of carriage of resistant organisms), and respiratory viral infections. Colonization is seasonal, peaking in the winter months but present in children year-round. Colonizing organisms can be transmitted from person to person by an aerosol route from coughing, with particles in the 1- to 5-µm size range depositing primarily in the upper respiratory tract62 or acquired from saliva and shared drinking vessels.63
The frequency of pneumococcal colonization can be influenced by the resident polymicrobial microbiome of co-colonizing organisms, which include other streptococci and Neisseria species, as well as Haemophilus influenzae, Moraxella catarrhalis, Staphylococcus aureus, and other organisms. These bacteria interact and can compete for nutrients and binding sites, produce inhibitory molecules, and modify both local innate and specific immune responses.60,64 Bacterial components can have specific interactions with host mucosal constituents.60,65 Pneumococcal neuraminidases (NanA and B) may modify neighboring bacteria and cleave sialic acid on host mucins and glycopeptides to facilitate exposure of N-acetylglucosamine (GlcNAc) receptors on resting epithelial cells that bind pneumococcal surface-associated proteins, such as pneumococcal surface adhesion protein A (PsaA). Local inflammation induced by, for instance, rhinovirus or influenza infections, and conditions that elicit proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1), facilitate pneumococcal binding, uptake, and likely migration of the bacteria across the epithelium and endothelium.64 Such inflammation elicits upregulation of epithelial platelet-activating factor receptor that binds bacterial cell wall phosphoryl choline (ChoP), which intercalates through the capsule. Moreover, sialic acid, lacto-N-neotetraose, and polymeric immunoglobulin receptors (pIgR) on activated epithelial cells bind surface-expressed CbpA to advance transcytosis.65
Other innate host macrophage receptors, including scavenger receptor A (SRA), mannose receptor, and particularly, macrophage receptor with collagenous structure (MARCO), have been proposed in murine models to enhance mucosal clearance of S. pneumoniae. The role of MARCO may be to enhance interactions between the organisms and other innate receptors, such as Toll-like receptor 2 (TLR2) and nucleotide oligomerization domain protein 2 (NOD2), to promote cytokine and chemokine production.66 In addition to its cytotoxic and complement-modifying activities, pneumolysin, largely conserved among strains, appears to engage TLR4, and, in conjunction with the cell wall, to limit mortality after mucosal challenge.67 Finally, specific host IgA bound to the pneumococcal capsule, and potentially to other surface proteins, can be cleaved by bacterial IgA1 protease. The bound but cleaved IgA modifies the surface charge and can enhance binding of encapsulated pneumococci that otherwise bind less well to the epithelium.68 Pneumococci themselves may also respond to local environmental conditions by increasing expression of ChoP and CbpA to enhance adherence to mammalian cells.65 Thus, S. pneumoniae adapt to and interact with their mucosal milieu to effect colonization, but the presence of inflammation may promote the conversion from asymptomatic colonization to local and systemic disease. When symptomatic infections do accompany colonization, they typically occur within a few weeks of acquiring a new strain rather than from chronic carriage, presumably because antibody production generally follows soon after colonization in children52 or in adults.69
Pneumococcal Capsular Serotypes and Progression from Colonization to Disease
The serotypes of colonizing pneumococci can vary by age and geography and are predictive of the incidence, syndrome, and outcome of pneumococcal infections. Only a relatively small number of the more than 92 capsular serotypes typically cause serious disease, particularly among unvaccinated children. Differences among these capsular types account for approximately 60-fold differences in the invasiveness of the organism.70–73 Indeed, capsular serotype appears to be a primary determinant of which strains are likely to remain as colonizers (e.g., 6A, 6B, 9N, 19F, and 23F), which are more likely to progress from carriage to cause more invasive disease (e.g., 14, 5, 1, 7F, 18C, 8, 38, and 33F), and, among those causing invasive disease, which are more likely to cause fatal infections.60,71,74–77 Based on a process called phase variation, organisms that express less capsule in vitro are more likely to colonize, whereas those producing more capsule show greater resistance to phagocytosis.78 The clinical distinctions among organisms are associated with their interaction with the host. Colonizing strains are proposed to resist nonopsonic phagocytosis but to be cleared by alveolar macrophages and by complement in blood and, perhaps, lung, thereby resulting in a lower incidence of invasive disease.79 In contrast, invasive serotypes are less common colonizers that, if not cleared by neutrophils, show increased adhesion-mediated binding to and translocation across the epithelium and are more resistant to killing by alveolar macrophages and complement. However, when colonizing strains do cause invasive disease (e.g., serotypes 3, 6A, 6B, 9N, 19F, and 23F), mortality is higher than with infection with the usual invasive strains, perhaps related to decreased host resistance.
Although the serotype distribution varies by age, these differences are likely not sufficient to explain the increases in mortality from (1) infancy to adulthood, (2) age 65 to 80 years to older than 80 years, or (3) in the presence of underlying disease among adults older than 65 years.31 Disease in immunocompromised adults is often caused by pediatric serotypes.80 Thus, the interaction between microbial factors, particularly expression of capsular serotype76 and the integrity of host defense31,77 determine the development of disease and mortality, but serotype appears to be the primary determinant of outcome in young healthy adults.
These morbid outcomes highlight the importance of understanding, potentially preventing, or at least successfully managing pneumococcal colonization and thereby its consequences. In this context, use of the polysaccharide-protein conjugate vaccines over the last decade has been associated with decreased rates of colonization in immunized children.81,82 This decrement in childhood colonization and disease has been accompanied by a progressive reduction in adult invasive disease with the pediatric vaccine serotypes.83 Colonization itself may serve as an immunizing event, and antibodies to pneumococcal neuraminidase (NanA), PspA, and capsular polysaccharides have been detected in serum after carriage.60,65,69,84 These antibodies, as well as antibody-independent CD4+ T cell-mediated responses,85 perhaps involving Th17 CD4+ T cells,79,86,87 may also mediate protection against subsequent colonization and the development of disease and provide alternative approaches to immunization.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


