Yok-Ai Que, Philippe Moreillon
Staphylococcus aureus (Including Staphylococcal Toxic Shock Syndrome)
Staphylococcus aureus is a highly successful opportunistic pathogen. It is a frequent colonizer of the skin and mucosa of humans and animals (it is present in the anterior nares of up to 30% of the healthy human population) and can produce a wide variety of diseases. These diseases encompass relatively benign skin infections, such as folliculitis and furunculosis, and life-threatening conditions, including erysipelas, deep-seated abscesses, osteomyelitis, pneumonia, sepsis, and endocarditis.1 In addition to infections in which the organism is physically present at the infected site, S. aureus is also capable of producing “distant” diseases, which are mediated by the secretion of toxins.2 The toxins can be produced directly by bacteria that colonize the skin or mucosa or indirectly by microorganisms that colonize food or beverages. The former is exemplified by staphylococcal scalded skin syndrome (SSSS),3 which is the result of mucosal or wound colonization by S. aureus–producing exfoliative toxin A or B (ETA or ETB) and by staphylococcal toxic shock syndrome (TSS),4 which is the result of the production of toxic shock syndrome toxin 1 (TSST-1) or exotoxins B or C. The latter is exemplified by S. aureus food intoxication, in which the toxin is ingested with the contaminated dish and disease follows shortly thereafter in the form of vomiting and diarrhea. Food intoxication is the result of staphylococcal toxins called enterotoxins.2,5 These toxins are heat stable. Cooking may kill the contaminants but does not denaturate the toxins. Hence, subsequent culture of the dish may fail to grow the culprit bacterium.
S. aureus has an extraordinary capacity to adapt and survive in a great variety of environments. During the past decades, molecular and genetic dissection of S. aureus has revealed a great number of surface adhesins, which mediate adherence to and colonization of target tissues, and secreted enzymes and toxins that are responsible for invasion and distant disease (Table 196-1).1,6,7,8 The availability of the complete nucleotide sequence of more than 41 S. aureus genomes (www.ncbi.nlm.nih.gov/genome/genomes/154) has helped complete this portrait. Approximately 50% of the S. aureus genome shares homology with notoriously nonpathogenic Bacillus subtilis, which indicates that the two organisms have evolved from a common ancestor.9,10 Homology searches on the chromosome revealed numerous new surface-attached and secreted factors that represent additional pathogenic factors. S. aureus harbors a large number of mobile genetic elements (MGEs) from exogenous origin, including insertion sequences, transposons, bacteriophages, pathogenicity islands, and genomic islands, which contain specific determinants responsible for disease and antibiotic resistance.6,7,10,11,12 The presence of these exogenous elements attests to the high capacity of S. aureus to undergo horizontal gene transfer and exchange genetic elements with other organisms, including staphylococcal and nonstaphylococcal genera. Because gene exchange is a key player of evolution, this peculiar genetic plasticity is a likely explanation for the success of S. aureus, both as a colonizer and a disease-producing microbe. In the case of superantigens (see subsequent discussion), one of the trading partners is suspected to be Streptococcus pyogenes.4
TABLE 196-1
Staphylococcus aureus Extracellular Factors Involved in Pathogenesis and Response to Global Regulatory Elements during Bacterial Growth
| GENE | LOCATION | PRODUCT | ACTIVITY/FUNCTION | TIMING* | ACTION OF REGULATORY GENES† | |||
| agr | saeRS | rot | sarA | |||||
| Surface Proteins | ||||||||
| spa | Chromosome | Protein A | Anti-immune, anti-PMN | exp | − | ‡ | + | |
| cna | Chromosome | Collagen BP | Collagen binding | exp | − | |||
| fnbA | Chromosome | Fibronectin BPA | Fibronectin binding | exp | − | + | ||
| fnbB | Chromosome | Fibronectin BPB | Fibronectin binding | exp | − | + | ||
| clfA | Chromosome | Clumping factor A | Fibrinogen binding | exp | 0 | |||
| clfB Chromosome | Chromosome | Clumping factor B | Fibrinogen binding | exp | 0 | + | 0 | |
| lfb | Chromosome | Lactoferrin BP | Lactoferrin binding | exp | ||||
| Capsular Polysaccharides | ||||||||
| cap5 | Chromosome | Polysaccharide capsule type 5 | Antiphagocytosis? | pxp | + | + | ||
| cap8 | Chromosome | Polysaccharide capsule type 8 | Antiphagocytosis? | pxp | + | |||
| Cytotoxins | ||||||||
| hla | Chromosome | α-Hemolysin | Hemolysin, cytotoxin | pxp | + | + | – | ‡ |
| hlb | Chromosome | β-Hemolysin | Hemolysin, cytotoxin | pxp | + | + | – | ‡ |
| hld | Chromosome | δ-Hemolysin | Hemolysin, cytotoxin | Pxp | + | 0 | + | |
| hlg | Chromosome | γ-Hemolysin | Hemolysin, cytotoxin | Pxp | + | – | ‡ | |
| lukS/F | PVL phage | PVL | Leucolysin | Pxp | + | – | ||
| Superantigens | ||||||||
| sea | Bacteriophage | Enterotoxin A | Food poisoning, TSS | Xp | 0 | |||
| seb | SaPI3§ | Enterotoxin B | Food poisoning, TSS | Pxp | + | ‡ | ||
| sec | SaPI4§ | Enterotoxin C | Food poisoning, TSS | Pxp | + | |||
| sed | Plasmid | Enterotoxin D | Food poisoning, TSS | Pxp | + | |||
| eta | ETA phage | Exfoliatin A | Scalded skin syndrome | Pxp | + | |||
| etb | Plasmid | Exfoliatin B | Scalded skin syndrome | Pxp | + | |||
| tst | SaPI1,2, bov1§ | Toxic shock toxin-1 | Toxic shock syndrome | Pxp | + | ‡ | ||
| Enzymes | ||||||||
| SplA-F | Chromosome | Serine protease-like | Putative protease | + | – | |||
| ssp | Chromosome | V8 protease | Spreading factor | Pxp | + | 0 | – | |
| aur | Chromosome | Metalloprotease (aureolysin) | Processing enzyme? | Pxp | + | – | ||
| sspB | Chromosome | Cysteine protease | Processing enzyme? | ? | – | |||
| scp | Chromosome | Staphopain (protease II) | Spreading, nutrition | Pxp | + | – | ||
| geh | Chromosome | Glycerol ester hydrolase | Spreading, nutrition | Pxp | + | 0 | – | ‡ |
| lip | Chromosome | Lipase (butyryl esterase) | Spreading, nutrition | Pxp | + | 0 | ‡ | |
| fme | Chromosome | FAME | Fatty acid esterification | Pxp | + | ‡ | ||
| plc | Chromosome | PI-phospholipase C | Pxp | + | ||||
| nuc | Chromosome | Nuclease | Nutrition | Pxp | + | + | ||
| has | Chromosome | Hyaluronidase | Spreading factor | Xp | ‡ | |||
| coa | Chromosome | Coagulase | Clotting, clot digestion | Exp | + | + | + | |
| sak | Bacteriophage | Staphylokinase | Plasminogen activator | Pxp | + | 0 | ||
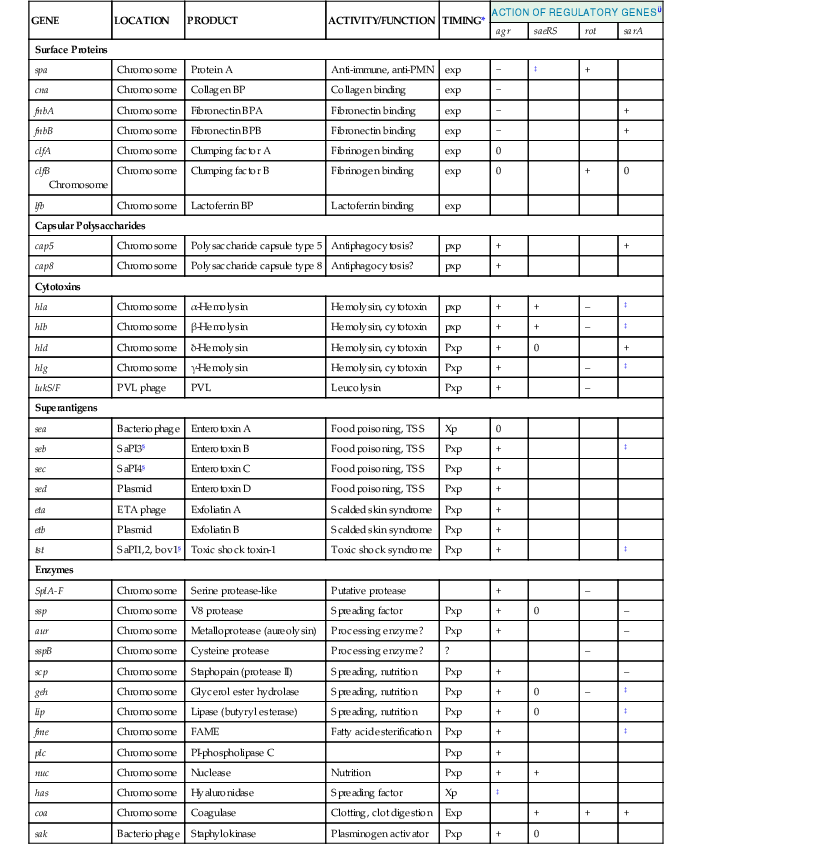
* Timing: xp, throughout exponential phase; exp, early exponential phase only; pxp, postexponential phase; 0, no effect of gene on. Expression: +, upregulated; –, downregulated.
† agr, accessory gene regulator; PVL, Panton-Valentine leukocidin; saeRS, S. aureus exoproteins; rot, repressor of toxins; sarA, Staphylococcus accessory regulator.
‡ Controversial.
§ SaPI, S. aureus pathogenic island.
BP, binding protein; FAME, fatty acid modifying enzyme; PMN, polymorphonuclear neutrophil.
Modified from Cheung AL, Projan SJ, Gresham H. The genomic aspect of virulence, sepsis, and resistance to killing mechanisms in Staphylococcus aureus. Curr Infect Dis Rep. 2002;4:400-410; and Novick RP, Geisinger E. Quorum sensing in staphylococci. Ann Rev Genet. 2008;42:541-564.
The Microorganism
Members of the Staphylococcus genus are gram-positive cocci (0.5 to 1.5 µm in diameter) that occur singly and in pairs, tetrads, short chains, and irregular grapelike clusters. Ogston13 introduced the name Staphylococcus (Gk. staphylé, “a bunch of grapes”) to describe micrococci responsible for inflammation and suppuration. Staphylococci are nonmotile, non–spore forming, and usually catalase positive, and they are often unencapsulated or have a limited capsule (Fig. 196-1). Most species are facultative anaerobes.9
The genus Staphylococcus contains up to 40 taxa, 16 of which are commonly found in humans (Table 196-2). Only a few are pathogenic in the absence of predisposing immunosuppression or implanted foreign material. The most virulent ones include S. aureus and S. lugdunensis in humans and S. aureus and S. intermedius in animals. Although S. epidermidis, S. haemolyticus, and S. saprophyticus are commonly responsible for device-related and urinary tract infections, they produce substantially less devastating disease syndromes than S. aureus.
TABLE 196-2
Some Staphylococcal Species from Mammals and Relationship between Production of Coagulase and Clumping Factor (Fibrinogen-Binding Protein A) and Potential Virulence
| HOST | SPECIES | COAGULASE* | CLUMPING FACTOR* | VIRULENCE* |
| Human and other primates | S. aureus | ++ | ++ | +++ |
| S. epidermidis | – | – | + | |
| S. capitis | – | – | ± | |
| S. caprae | – | – | ± | |
| S. saccharolyticus | ± | – | – | |
| S. warneri | – | – | – | |
| S. pasteuri | – | – | – | |
| S. haemolyticus | – | – | + | |
| S. hominis | – | – | ± | |
| S. lugdunensis | – | ± | + | |
| S. auricularis | – | – | ± | |
| S. saprophyticus | – | – | + | |
| S. cohnii | – | – | – | |
| S. xilosus | – | – | – | |
| S. simulans | – | – | – | |
| S. schleiferi | ± | + | + | |
| Carnivores | S. intermedius | + | – | ++ |
| S. felis | – | – | ++ |
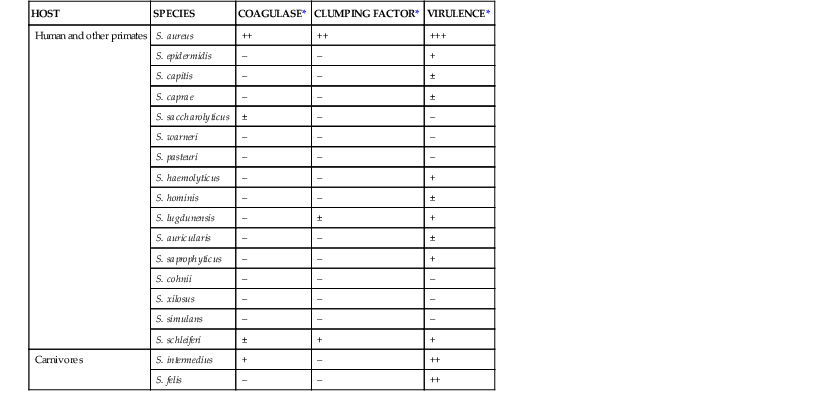
* Semiquantitative estimate of production of coagulase and clumping factor and relation to virulence.
Modified from Kloos WE, Schleifer KH, Goetz F. The genus Staphylococcus. In: Balows A, Trüper HG, Dworkin M, et al, eds. The Prokaryotes. 2nd ed. New York: Springer-Verlag; 1992:1369-1420; and Kloos WE, Bannerman TL. Staphylococcus and Micrococcus. In: Murray PR, Baron EJ, Pfaller MA, et al, eds. Manual of Clinical Microbiology. 6th ed. Washington, DC: ASM Press; 1995:282-298.
S. aureus harbors some unique features when compared with its less disease-producing congeners. These include coagulase and clumping factors (or fibrinogen-binding proteins), which have laboratory diagnostic value because they help rapidly discriminate between coagulase-positive (i.e., S. aureus) and coagulase-negative staphylococci (CoNS; see Table 196-2). Moreover, S. aureus carries between more than 20 and more than 30 adhesin genes and toxin genes, respectively, as compared with 10 or less adhesin genes and no toxin genes for the CoNS mentioned previously.7,14–16 Thus, S. aureus is a distinct pathogen within the Staphylococcus genus.
Habitat
Staphylococci are ubiquitous colonizers of the skin and mucosa of virtually all animals, including mammals and birds.9 Some species have preferential niches as indicated by their names (see Table 196-2). S. epidermidis and S. capitis are constant colonizers of the skin and scalp, respectively.
In animals S. aureus is a major cause of livestock infection, including mastitis in bovine and ovine herds. In humans, S. aureus has a niche preference for the anterior nares, especially in adults,17,18 and is shed onto healthy skin, including axilla and perineum. It can exist as a resident or a transient member of the normal flora. Nasal carrier rate varies from 10% to 40% in both the community and the hospital environment. Chronic nasal carriage may put certain populations at an increased risk for infection, such as patients with recurring furunculosis and patients who are subject to medical procedures, including hemodialysis, peritoneal dialysis, and surgery (see later section “Carriage of S. aureus” under “Clinical Aspects and Epidemiology”).19,20
Nasal carriage of S. aureus has also become a way of persistence and spread of multiresistant staphylococci, especially methicillin-resistant S. aureus (MRSA).17,18 Because MRSA can resist practically all available antibiotics, it has risen to the level of public health threat, in the hospital for 3 decades and in the community since the beginning of this century.21,22
Culture and Identification
Live organisms obtained via cultures are critical for phenotypic diagnosis and revealing emerging antibiotic resistances from as yet unknown mechanisms. In addition, molecular diagnosis helps speed up the results, which take a few hours instead of 1 to 3 days with bacterial subculturing. Molecular methods also help detect the presence of nonculturable microbes, mostly when patients have taken antibiotics before sample collection.
Techniques for culture and identification of staphylococci have been described.9 Specimens should be inoculated both on blood agar and into rich liquid media such as Mueller-Hinton broth. With S. aureus, abundant growth occurs normally within 18 to 24 hours. However, morphology variants (see subsequent discussion) may require prolonged growth periods, and plates should be kept 2 to 3 days to detect them. Colonies should be Gram stained, subcultured, and tested for genus, species, and antibiotic susceptibility when appropriate. Phenotypic tests for species identification include coagulase tests and agglutination tests, which detect the presence of surface determinants, including clumping factor, protein A, and polysaccharides.23 Molecular specification with polymerase chain reaction (PCR) amplification of the 16S or 23S ribosomal RNA (rRNA) may be necessary in case of unclear phenotypes. Currently, more rapid identification by matrix-assisted laser desorption ionization/time-of-flight (MALDI-TOF) mass spectrometry is being developed.24 Antibiotic resistance tests vary from agar-diffusion methods (e.g., Kirby-Bauer and Etests) to automated measurement of metabolic activity or growth rates. Macro broth or agar dilution methods are precise but are not routinely performed in the laboratory.25
Morphology Variants
Prolonged incubation is particularly important in detection of morphology variants such as small colony variants (SCVs). SCVs grow into tiny colonies that are difficult to distinguish and may be mistakenly disregarded as contaminants.26 They are usually recovered from protracted, difficult-to-treat infections such as chronic osteomyelitis and infected osteosynthetic prostheses and were recently described in patients with cystic fibrosis.27
The most classic types of SCVs are selected during aminoglycoside therapy and result from alterations in the respiratory chain. Such SCVs have a lower transmembrane potential, which impedes the intake of the drug.28 Interestingly, switching from normal colonies to SCVs occurs naturally in the absence of antibiotic at a high rate (about 10−6), and switching back from SCVs to normal colonies occurs as well.29 Hence, SCVs are proposed to result from an intrinsic capacity of the bacterium to survive rather than fortuitous mutations.
SCVs are also selected by other antimicrobial agents, including triclosan,30 and are recovered from the sputa of up to 20% of patients with cystic fibrosis carrying S. aureus treated with trimethoprim-sulfamethoxazole.27 These SCVs carry mutations in the thymidylate synthase gene (thyA) and are dependent on exogenous thymidine to grow.31 S. aureus synthesizes thymidine by using thyA plus tetrahydrofolate to convert uridine monophosphate into thymidine monophosphate. Trimethoprim inhibits the synthesis of tetrahydrofolate, thus making thyA useless. By mutating the thyA gene, S. aureus forces itself to rely on exogenous vital thymidine by importing it. This makes the bacterium resistant to trimethoprim. Thymidine is available in DNA-rich lung secretions of patients with cystic fibrosis as well as in abscesses. However, the rate of thymidine import is limiting, which results in slow growth and SCV phenotype.
In spite of slow growth, SCVs are equally or more infective than their fast-growing parents in experimental osteoarthritis32 and experimental endocarditis.33 Moreover, SCVs are particularly prone to invade eukaryotic cells and persist in them,26,34 which may explain their occurrence in latent infections. SCVs are cross-resistant to drug-induced killing by other antibiotics,27 and their eradication necessitates prolonged antibiotic therapy including drug combinations with rifampin.
Molecular Diagnosis
Molecular diagnosis plays an increasing role in rapid detection of microbial pathogens and identification of drug-resistance determinants. Results can be obtained within a few hours. Identification techniques based on molecular probing were recently reviewed.35 One of these techniques relies on fluorescent detection of 16S rRNA with a peptide nucleic acid probe (peptide nucleic acid fluorescence in situ hybridization [PNA-FISH]). Such a technique recently identified S. aureus in positive blood cultures and differentiated it from other staphylococci in 4 hours with 100% sensitivity and specificity.36
In addition, multiplex real-time PCR assay is being developed to quantify organisms directly in clinical samples. Genes representative of both species and resistance mechanisms are amplified simultaneously. For MRSA, the resistance gene sought is mecA, which encodes low-affinity penicillin-binding protein 2A (PBP2A). However, mecA is also present in methicillin-resistant CoNS and thus detects simultaneously both MRSA and commensal methicillin-resistant CoNS. Two studies showed that simultaneous amplification of additional regions that were specific for S. aureus could circumvent the problem and was successfully applied to screen MRSA directly from nasal samples.37,38 In one of these studies, one PCR primer was in mecA, which is present in both MRSA and methicillin-resistant CoNS, and the other targeted orfX, which is situated farther downward and is specific for S. aureus.37 In the other, the amplicons included mecA and two versions of femA specific for S. aureus and S. epidermidis, respectively.38
A wealth of newer molecular techniques are being developed for rapid differentiation between staphylococcal species and identification of major antibiotic-resistance mechanisms. These include ultra-high-throughput genome sequencing, RNomics, and proteomics.
Molecular Typing
S. aureus is common both in the hospital and in the community. In a survey of 3,309,413 microbial isolates recovered from sterile body sites in 300 U.S. laboratories (between 1998 and 2005), S. aureus was the most prevalent bacterium in inpatients and the second most prevalent isolate in outpatients (18.7% and 14.7% of the isolates, respectively).39 Moreover, the frequency of MRSA is as high as 59% in intensive care unit (ICU) patients, 55% in non-ICU patients, and 48% in outpatients.22,39 Because MRSA is of particular medical concern, major efforts were devoted to understand its evolution and population structure.40,41 MRSA is highly clonal, and a few highly successful clones, named according to the place where they were described, can be recovered at multiple locations both nationwide and worldwide (i.e., the Iberian, Brazilian, Hungarian, New York/Japan, Pediatric, and EMRSA-16 pandemic clones).40 The main typing methods underlying this comprehension are briefly presented subsequently. More ancient phage typing and ongoing methods using whole-genome sequencing are not addressed.42
Pulsed-Field Gel Electrophoresis
The most widely used method is a restriction-fragment length technique based on large chromosomal fragments generated by digestion with the low-frequency cutting enzyme SmaI. The fragments are separated with pulsed-field gel electrophoresis (PFGE) and yield banding patterns specific for particular clones. Banding comparison allowed identification of the major epidemic clones listed earlier, which represented 70% of more than 3000 MRSA isolates recovered worldwide.40
One limitation of PFGE is that it does not provide accurate information on the genealogy of the organism. Indeed, the length of chromosomal fragments, and thus the clone-specific banding, may be modified with acquisition or loss of mobile DNA (MGEs) such as transposons, prophages, or pathogenicity islands. The new banding pattern may identify a different clone, which is in fact the same bacterium that has gained or lost MGEs. The new organism may have acquired properties important for successful spread, but phylogenic relation to its parent remains.
Multilocus Sequence Typing
In contrast to PFGE, multilocus sequence typing (MLST) is a sequence-based method that allows the unambiguous assignment of the ancestral phylogeny of the staphylococcal population.43 It consists of sequencing seven housekeeping genes (i.e., arcC, aroE, glpF, gmk, pta, tpi, and yqiL) and comparing them with the sequences of other isolates collected in a central database (www.mls.net). It compares allelic diversity based on approximately 500-bp internal gene fragments. Thousands of sequences have been submitted, generating numerous sequence types (STs). Organisms that share all seven alleles are defined as clones, those that share five of seven identical alleles are defined as clonal complexes (CC), and those that share less than seven alleles are defined as unrelated.
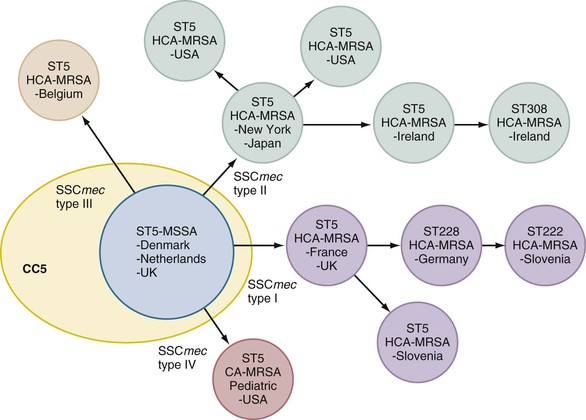
Because housekeeping genes are independent of acquired MGEs, MLST traces staphylococci back to their latest common ancestor. Of the seven pandemic clones mentioned previously, six could be traced back to three ancestral methicillin-susceptible S. aureus (MSSA; i.e., CC5, CC8, and CC30; Fig. 196-2).44 Hence, MLST indicates that a few ancestral clones of MSSA already took the lead and successfully colonized humans and animals before antibiotic resistance. Later acquisition of MGEs carrying drug-resistance or virulence genes helped further adaptation to new conditions (e.g., antibiotic use in hospitals), generating a new PFGE makeup on similar ancestral parents (see “Genomics and Evolution”).
Spa Typing and Double-Locus Spa-Clfb Typing
These sequence-based methods, spa typing and double-locus spa-clfB typing, rely on PCR amplification of strain-specific regions within hypervariable segments of the spa (protein A) or clfB (clumping factor B) genes.45 The variable regions are made of 24 nucleotide repeats in spa and serine-aspartate (SD) repeats in clfB, the length of which may vary from duplication or accidental loss of DNA material. These methods generate unambiguous datasets that can be compared in multicenter studies.
Typing is critical to understand the S. aureus epidemiology. As yet, however, no specific types could be attributed to disease-producing versus mere colonizing strains.11,46
Pathogenesis
Regulation and Virulence Determinants
S. aureus is extremely well equipped in surface factors and secreted proteins that mediate host colonization and disease (see Table 196-1).1 In addition to these features, S. aureus is equipped with regulatory systems that sense environmental conditions and respond by fine tuning the expression of given metabolic and virulence determinants (for review see articles by Cheung and colleagues,8 Novick,47 and Pragman and Schlievert48). Some aspects of this subtle adaptation machinery are described subsequently.
Regulation
At least three families of regulatory elements intertwine to adjust gene expression to specific environmental conditions: first, two-component regulatory systems, of which agr is a paradigm; second, DNA-binding proteins, largely represented by the Sar family of proteins; and third, small regulatory RNAs.
agr and Other Two-Component Regulatory Systems
The paradigm of two-component regulatory systems virulence gene regulation is agr, which stands for accessory gene regulator and is schematized in Figure 196-3.49 agr functions as a quorum sensing control that reacts to bacterial density, allowing the preferential expression of surface adhesins during the exponential phase of growth (low cell density) and switching to the expression of exoproteins during the postexponential and stationary growth phases (high cell density).47,49 The switch is composed of two divergent operons (see Fig. 196-3). On the left hand, promoter P2 drives the transcription of a series of components that comprises (1) a transmembrane protein (AgrB); (2) an autoinducing peptide precursor (AgrD), which is processed and exported by membrane-spanning AgrB; (3) a transmembrane sensor (AgrC), which is the cognate receptor of the AgrD-derived autoinducing peptide; and (4) a transcription regulator (AgrA) that can be activated by AgrC. At low cell density (exponential growth phase), the P2 promoter is off and the operon is transcribed at a low level. As cell growth proceeds, the concentrations of both bacteria and extracellular autoinducing peptide increase in the milieu, thereby augmenting the chance of the autoinducing peptide to make contact with its cognate AgrC receptor. On contact, AgrC activates the response regulator AgrA, a process that may involve AgrA dephosphorylation.47
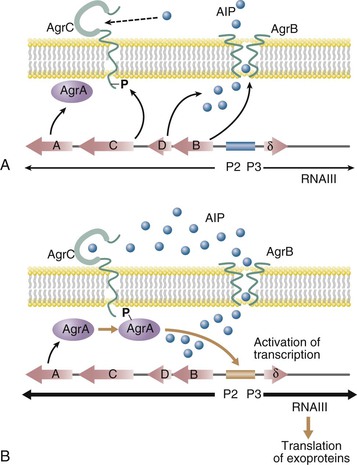
Activated AgrA is a DNA-binding protein that turns on the transcription from both promoter P2, generating a positive feedback on the system, and promoter P3, which drives the transcription of δ-hemolysin and of a peculiar effector called RNAIII. RNAIII has a reciprocal effect and activates the expression of several secreted proteins while downregulating the expression of surface-bound factors (see Table 196-1). RNAIII has a complex three-dimensional structure and a long half-life (up to 15 minutes). It regulates gene expression in several ways, including at the translational level by blocking the messenger RNA (mRNA) ribosome-binding site of the target genes.
The S. aureus chromosome encodes for more than 10 two-component regulatory systems involved in both metabolic environmental control and virulence gene regulation. Important two-component regulatory systems regarding virulence genes include saeR/S (for S. aureus exoproteins),50 srrAB (for staphylococcal respiratory response),51 and arlS (for autolysis-related locus sensor).52 saeR/S was identified with transposon mutation in a pleiotropic mutant defective in exoprotein synthesis other than that regulated by agr (e.g., coagulase and nuclease; see Table 196-1).53 saeR/S acts independently of agr and responds to environmental stimuli such as high salt, low pH, glucose, and subinhibitory antibiotic concentrations. srrAB and arlS interfere with growth in microaerobic conditions and autolysis, respectively. srrAB represses the expression of toxic shock syndrome toxin-1 (TSST-1) and protein A in microaerobic conditions,51 an observation that may be relevant for the pathogenesis of tampon-related toxic shock syndrome (see subsequent discussion).4 Both srrAB and arlS interact reciprocally with agr.48,54
DNA-Binding Proteins
sar stands for staphylococcal accessory regulator.54 It is an important locus that encodes a DNA-binding protein, SarA, which positively controls agr (Fig. 196-4)8 and maybe also sae and arlS.47,54 In addition, sar directly regulates adhesin genes (see Table 196-1). The sarA transcripts peak at the end of the logarithmic phase of growth, thus promoting agr expression. Moreover, sarA itself is transcribed downstream of three alternate promoters, which can themselves be regulated by as yet incompletely solved factors.
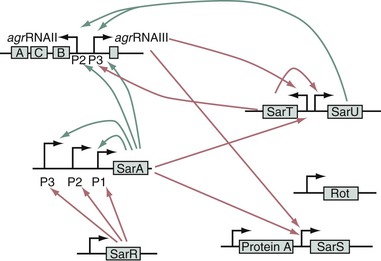
SarA is the prototype of a growing family of DNA-binding proteins that may drive a number of transcriptional activities, including the expression of housekeeping genes and phage-related genes. sarA homologues include sarR, sarS, sarT, sarV, sarU, sarY, and rot.48,54 rot stands for repressor of toxins and counters toxin expression by repressing agr.55 Inactivation of rot partially restored the agr phenotype in agr-negative mutants, probably by alleviating a repressing effect on the downstream P3 cascade of the agr. This downstream cascade might be the target of several additional regulators that also affect the agr phenotype (see Fig. 196-4).
Sigma factors (σ) are another major mechanism of response to environmental stimuli. In bacteria, σ factors combine with and activate RNA polymerase to transcribe specific sets of genes. S. aureus contains one σA and two alternative σB and σC. Alternative σB is important for the microbial response to a variety of stresses, including temperature, energy depletion, and chemical stimuli.56 It acts mostly via the global regulatory network but also has some direct effect by activating the expression of coagulase and fibronectin-binding proteins at the early growth phase and downregulating certain secreted proteins in the stationary phase. Mutants overexpressing σB were more virulent in experimental endocarditis, probably by increasing the expression of surface adhesins.57 But the pathogenic role of σB was not shown in other models.
Small RNAs and Endoribonuclease III
Small RNAs (sRNA) are increasingly recognized as major players in regulation of gene expression. They act mainly at the translational level via antisense hybridization with mRNA, where they can either alter mRNA stability, hide ribosome-binding sites (RBS) from ribosome recognition, or conversely reveal RBS that are hidden in secondary mRNA structures by unfolding these very structures. Alternatively, sRNA can also bind regulatory DNA-binding proteins, thus sequestering them from their original gene regulatory function. At least 12 regulatory sRNA have been identified in S. aureus,58 but many more are likely to come. The most prominent of them are RNAIII, which orchestrates the agr response, and RNAI, which regulates the replication of multiresistance plasmid pSK41.
In symmetry, post-transcriptional expression is also modulated by direct RNA alteration via endoribonuclease III (RNase III). This RNA double-stranded endonuclease plays a critical role in RNA processing and decay. It was recently shown to modulate post-transcriptional expression by various mechanisms, including turnover of transcribed and nontranscribed RNAs, as well as maturating the 5′ untranslated region (5′UTR) of the mRNAs of the cold-shock protein cpsA and maybe the protein A spa genes, to increase their stability and translation.59
This intricate network underlines the subtlety of the bacterial response to environmental stresses. The regulatory network must be considered as a metabolic hub that integrates both external and internal information and responds in the most appropriate way. Some of these circuitries are likely to complement each other. Interruption of one of them may cause compensation by others, thus introducing biases in the observed phenotype. In this complex system, agr appears to be a central switch toward which many other regulators converge (see Fig. 196-4).
Role in Pathogenesis
The intuitive agr-based model suggests that scattered growing bacteria produce primarily adhesins, promoting tissue colonization, whereas installed organisms that form dense populations switch to the production of hydrolytic enzymes and toxins for the purpose of feeding and escaping host defenses.8,47 Accordingly, inactivation of the function of agr alone decreased pathogenicity in experimental models of tissue destruction (e.g., subcutaneous abscesses), where exoprotein production is likely to be important.60 On the other hand, agr inactivation did not influence much the course of experimental endocarditis, where bacterial surface adhesins are critical for valve colonization.61 Indeed, although agr-negative mutants are hampered in exoprotein production, they are still fully equipped with surface-bound colonizing determinants (see Table 196-1). In contrast, inactivation of sar decreased infectivity in experimental endocarditis61 because in addition to its effect on agr expression (see Table 196-1; see Fig. 196-3), sar also acts directly on expression of surface-bound fibronectin-binding protein A, which promotes experimental endocarditis.62,63
In addition, in vivo gene expression revealed a further level of complexity.48 For instance, although sar transcripts were detected in infected vegetations during experimental endocarditis, they were expressed from both P1 and P2 promoters, rather than only from the P1 promoter as observed in vitro.64 Likewise, in vivo expression of several genes appeared dissociated from their control regulator as described in vitro. Although agr positively regulates TSST-1 in vitro (see Table 196-1), the toxin was still expressed by an agr-negative mutant in a rabbit model of toxic shock syndrome in vivo.65 This may result from alternative regulation by other regulators that act either downstream of the agr locus or directly on the tss gene promoter. Eventually, agr-negative mutants can be recovered from clinical samples. Such agr-negative clinical isolates, and agr-negative laboratory mutants, have an increased ability to form biofilms, which are found in chronic infections such as osteomyelitis and device infections.66
Hence, the pathogenic implication of regulatory circuitries cannot be drawn merely from in vitro observations. In vivo experimentation reveals the plurality of S. aureus infection forms, which may be variously altered by novel antivirulence therapies. For instance, inhibition of the agr loop by acting on the autoinducing peptide impedes acute tissue destruction60 but might promote biofilm formation and chronic infection.
Ecologic and Epidemiologic Implication of agr
Genetic and functional experiments revealed the existence of at least four agr groups in S. aureus, which were characterized by specific variations in all three AgrB, AgrD, and AgrC proteins (Fig. 196-5).67 Whereas the autoinducing peptide of a given agr group stimulated signaling in other strains sharing the same agr group, it either cross-inhibited (e.g., group I and group IV) or cross-activated (e.g., group I and group II) members of other groups. This suggests that certain antagonistic agr groups could be mutually exclusive with attempts to simultaneously colonize the same niche. However, studies regarding this hypothesis gave conflicting results. In particular, patients with cystic fibrosis colonized with S. aureus can successfully harbor organisms from two antagonistic agr groups.
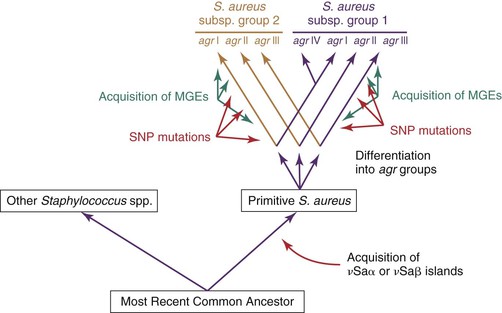
Although agr and other global regulators control the timely expression of pathogenic genes, they are not bona fide pathogenic factors themselves. The agr locus has homologues in numerous nonpathogenic staphylococci. A phylogenic study of nonpathogenic CoNS indicated that variations in agr genes followed parallel variations in species-specific rRNA genes.68 In fact, agr groups diverged very early on during the evolution of staphylococci (see “Comparative Genomics and Evolution”) and represent a lineage marker of strains that evolve in distinct environments rather than strategy to exclude potential competitors. Thus, global regulators were originally meant to control the expression of useful metabolic genes. How exogenous virulent genes, which were acquired later, succeeded in taking advantage of such systems remains a fascinating question of evolutionary genetics.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


