reversible, oxygen-linked changes in the rheologic properties of the sickle erythrocyte that characterizes the disease. Upon oxygenation, these polymers dissolve or “melt,” and the sickle erythrocyte loses most of those pathologic properties caused by the presence of polymer. If the concentration of Hb S in such solutions or in the red cell approaches 30 g/dl, a semisolid gel forms.
TABLE 33.1 WORLDWIDE PREVALENCE OF SICKLE CELL TRAIT AND DISEASE | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Hb, thereby inducing sickling. In a study of cultured human red cells and mice, the induction of adenosine A(2B) receptor led to increased levels of 2,3-diphosphoglycerate in the cells, consequently inducing more sickling.38 Recent findings point to the role of free heme in the kinetics of sickling; increasing amounts of free heme added to dialyzed Hb S solutions enhances polymerization.39
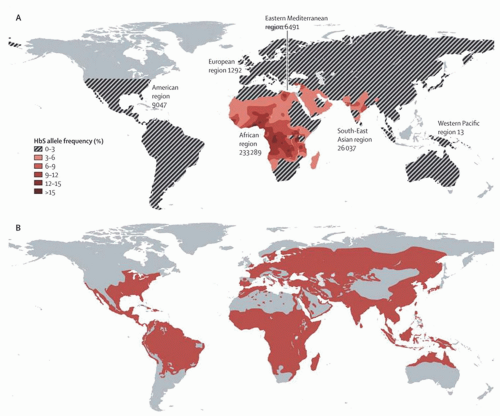 FIGURE 33.1. Global distributions of Hb S and malaria. This map shows the distribution of the Hb S allele. Part A indicates estimates for the combined yearly total number of individuals affected by Hb SS, Hb SC, and Hb Sβ-thalassemia by World Health Organization region. Part B shows the global distribution of malaria (red) before intervention to control malaria. From Rees DC, Williams TN, and Gladwin MT. Lancet. 2010 Dec 11;376(9757):2018-2031, with permission. |
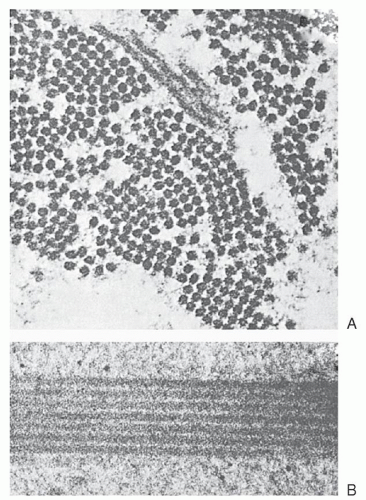 FIGURE 33.2. Electron photomicrographs of cell-free pellets of deoxyhemoglobin (Hb) S. A: Transverse section through fibers of polymerized Hb S (×97,000). B: Longitudinal section through same (×102,000). From Finch JT, Perutz MF, Bertles JF, et al. Structure of sickled erythrocytes and of sickle-cell hemoglobin fibers. Proc Natl Acad Sci U S A 1973;70:718, with permission. |
These conditions/factors also cause increased adhesion of sickle cells to endothelium in vitro. Thrombospondin may be an important plasma adhesogen because of its ability to bridge CD36 expressed on sickle reticulocytes.74 Thrombospondin-mediated adherence occurs under flow conditions with both microvascular and large-vessel endothelial cell assays. Thrombospondin levels are elevated in sickle cell patients during crisis,75 perhaps as a result of platelet activation. Fibronectin may link endothelial receptors with the fibronectin receptor a4 b1 (VLA-4) expressed on sickle reticulocytes.76, 77 Ultralarge forms of von Willebrand factor are postulated to promote adherence through nonreceptor mechanisms such as by binding with co-clustered hemichrome-band 3 aggregates on sickle membranes.78 Erythrocyte membrane sulfatide, a sulfated glycosphingolipid, may also play an important role in adhesion and its blockage with specific antibodies significantly prevents endothelial adhesion.79 Different mechanisms may predominate under various circumstances or in different parts of the circulation. Coagulopathy might cause thrombospondin release and precipitate vaso-occlusion in microvessels, and dehydration-induced vasopressin elevation might stimulate von Willebrand factor release and precipitate vaso-occlusion in large postcapillary venules. The increased sickle cell adherence to injured endothelium, the role of platelet and leukocyte receptors, and the possible stimulation of nitric oxide (NO) production by adherent red cells all require further evaluation.
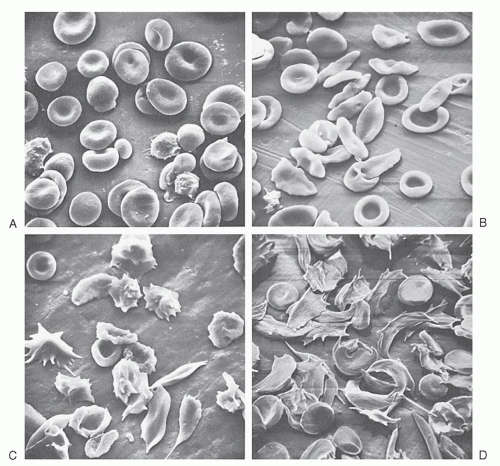 FIGURE 33.3. Erythrocytes from a patient with sickle cell anemia (SCA) examined with scanning electron microscopy. A: Oxygenated blood. Red cells appear normal except for one microspherocyte. Three leukocytes are evident in the field. B: Oxygenated irreversibly sickled cells are smooth in texture and outline but are ovoid or boatlike in shape. C: Partial deoxygenation causes the cells to assume bizarre shapes with spikes, spicules, and filaments that protrude from the cells. D: More complete deoxygenation causes the cells to assume sickled shapes with longitudinal surface striations. |
contribute to altered blood flow depends in part on the method used to study the properties of sickle red cells. Reduction in the cellular deformability of oxygenated sickle red cells has been demonstrated by increased viscosity of sickle blood (viscometry),83 decreased filtration of dilute cell suspensions through narrow pores,87 decreased ability of cells to undergo deformation in shear fields (ektacytometry),88 and increased aspiration pressures needed to induce entry of cells into micropipets.89 Cellular dehydration, as well as the resulting increase in cytoplasmic viscosity, is a major determinant of abnormal rheologic behavior of oxygenated sickled red cells. Sickled red cell membranes demonstrate extensional rigidity and persistent deformation, as documented by videomicrographs of micropipet aspiration.89 The rheologic properties of oxygenated sickle cells are strongly influenced by the state of cell hydration and the increased propensity for oxidative damage to the membrane.90, 91 The already compromised deformability of oxygenated sickle cells is dramatically reduced further after deoxygenation. Under conditions of high shear stress, increased internal viscosity appears to determine the rheologic behavior of ISCs, whereas at low shear rates, membrane rigidity assumes greater significance.92 Under physiologic conditions, increased viscosity results primarily from cellular dehydration. The poor deformability of ISCs, as measured by ektacytometry, can be rectified by osmotically hydrating them to a normal MCHC. The membrane rigidity of oxygenated sickle cells also can be returned to a normal level by replacing Hb S with Hb A, suggesting that the interaction of Hb S with the cell membrane is an important determinant of cellular rigidity.93 Peripheral vascular resistance is increased in proportion to ISC numbers, the extent of ISC deoxygenation, and ISC density.94 The functional significance of the impaired flow properties of sickle red cells has been demonstrated by measuring exercise tolerance before and after partial exchange transfusion. By increasing the relative number of cells containing Hb A without increasing the total Hb concentration, exercise capacity improved significantly.95
fragment of cross-linked fibrin, increases during vaso-occlusive crises and returns to normal after crisis resolution.170 Platelet activation is profoundly inhibited by NO, and this inhibition is blocked by plasma hemoglobin-mediated NO scavenging.171 Whether these alterations in hemostasis and fibrinolysis are of pathogenic significance or are simply epiphenomena remains to be determined.
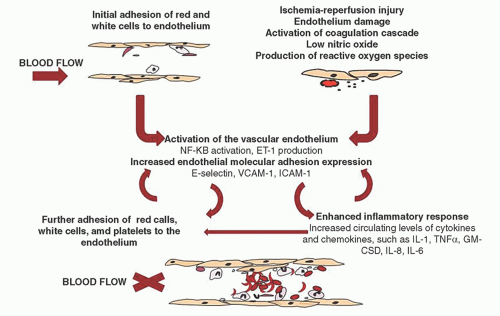 FIGURE 33.4. The chronic inflammatory state and vaso-occlusion. Hemoglobin S polymer formation leads red blood cell membrane surface exposure of glycolipids and protein epitopes. Red and white cell adhesion to the endothelium, coupled with endothelial damage due to cell-free hemoglobin and ischemia reperfusion injury, low nitric oxide bioavailability, and activation of the coagulation cascade by glycolipds, lead to activation of the vascular endothelium. Endothelial activation augments NFκB activity and endothelin-1 production, in association with increased surface adhesion molecule expression. Further adhesive interactions between the endothelium and red cells, leukocytes, and platelets are induced, coupled with a pancellular activation that results in an up-regulation of numerous inflammatory mediators. As such, a vicious circle of repeated cell activation, cellular adhesion, and inflammatory molecule production perpetuates the chronic inflammatory state that has a fundamental role in the vaso-occlusive process. Modified from Conran N, Franco-Penteado CF, Costa FF. Hemoglobin. 2009;33(1):1-16. |
microcirculation by intravascular sickling. Modest exacerbation of anemia and increased leukocytosis are common. Infections often precede vaso-occlusive episodes in children, suggesting that fever, dehydration, and acidosis may be contributing factors. In adults, a triggering event is not often identified, however.
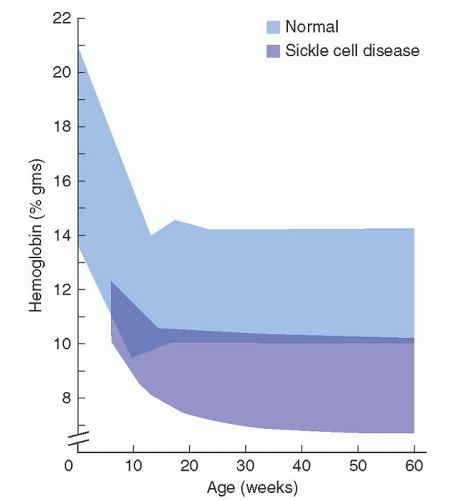 FIGURE 33.5. Hemoglobin concentration as a function of age in infants with sickle cell anemia (SCA). From O’Brien RT, McIntosh S, Aspnes GT, et al. Prospective study of sickle cell anemia in infancy. J Pediatr 1976;89:205, with permission. |
failed to demonstrate reduction of painful crises.206, 207 Despite encouraging results in Phase II trials, a prospective, multicenter, double-blind, randomized, placebo-controlled clinical trial for up to 72 hours of inhaled NO gas failed to demonstrate benefit in reducing duration of pain crisis or amount of narcotic use.208 Vitamin D levels are reported low in the majority of individuals with sickle cell disease, and there are reports linking chronic pain with vitamin D insufficiency and deficiency.209, 210, 211, 212 Replacement of vitamin D has been reported in a few patients to improve symptoms of chronic pain.213, 214 The duration and severity of pain crises are notoriously variable, and the natural course is one of spontaneous improvement. Consequently, uncontrolled reports of effective therapies must be viewed with skepticism, especially if the proposed treatment entails an element of risk.
cell adhesion, coagulation, hemostasis, cell proliferation, oxidative biology, and other functions. In 92 patients who had had an overt stroke, the network was used to determine 11 genes whose variants had a direct effect modulated by hemoglobin F levels and 9 genes whose variants were indirectly associated with stroke.
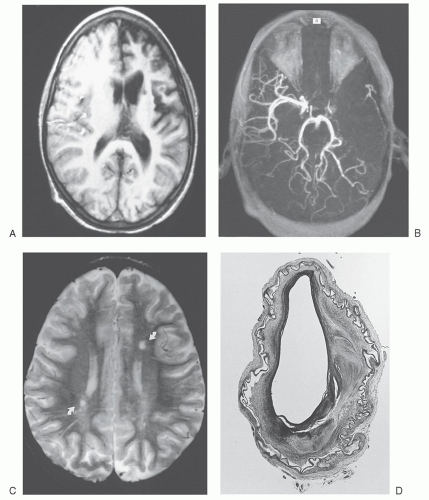 FIGURE 33.6. Vaso-occlusive effects in the central nervous system. A: T1-weighted magnetic resonance imaging (MRI) in a 6-year-old girl with hemoglobin (Hb) SS and a history of stroke. There is extensive atrophy involving the distributions of the left anterior and middle cerebral arteries with compensatory enlargement of the left lateral ventricle. B: Magnetic resonance angiography in the same patient showing occlusion of left middle cerebral artery and diminished flow through both anterior cerebral arteries. C: T2-weighted sagittal MRI in a 4-year-old boy with “silent infarcts.” Small areas of leukomalacia are seen in deep white matter in frontal and parietal areas (arrows). D: Pathologic section of internal carotid artery showing fibrinous thrombus with parallel layers of fibrin deposited on intimal surfaces and atrophic media. Courtesy of Dr. J. J. Jenkins, St. Jude Children’s Research Hospital. |
discontinued when they reach adulthood. In one series of nine adult patients who stopped transfusion after a median of 6 years, none suffered recurrent stroke.258 In an initial report, long-term treatment with hydroxyurea appeared to prevent stroke recurrence in children who were treated with relatively high doses (30 to 40 mg/kg/day).259, 260 In a larger prospective trial, 35 children with SCA and stroke had transfusions discontinued and hydroxyurea started.261 Initially, transfusion was stopped before hydroxyurea therapy was started, but later, transfusion was overlapped until full-dose hydroxyurea therapy was tolerated. Overall, stroke reoccurred at a rate of 5.7 events per 100 patient-years, but children receiving overlapping therapy had only 3.6 events per 100 patient-years. In a cohort from Jamaica, where blood supply is limited, hydroxyurea therapy was the only reliable treatment option and was shown to prevent stroke recurrence in children with prior stroke events.262 These findings led to the Stroke with Transfusions Changing to Hydroxyurea (SWiTCH) study, a phase III randomized, multicenter clinical trial, which compared standard treatment (transfusions with iron chelation) to alternative treatment (hydroxyurea with phlebotomy) for stroke prophylaxis in children who had experienced prior stroke.263, 264, 265 SWiTCH was a noninferiority trial with a composite primary endpoint of stroke and hepatic iron overload. This study was interrupted early because more strokes were observed in the hydroxyurea/phlebotomy arm and only equivalence in liver iron content was seen in the two arms. Chronic transfusion therapy and iron chelation remains the standard treatment for secondary prevention of stroke in children with SCA. Unfortunately, chronic blood transfusion does not prevent progression of brain vasculopathy or silent cerebral infarcts in children receiving this therapy for secondary stroke prevention, which makes blood transfusion an imperfect choice for this indication.263, 264
highest in children under 10 years of age.282, 283 Currently, conditional TCD velocity is not an indication for treatment, but the ongoing phase III multicenter Sparing Conversion to Abnormal TCD elevation (SCATE) study is investigating whether hydroxyurea therapy in children under age 11 years with conditional TCD will be less likely to develop abnormal TCD velocities than those who are observed (clinicaltrials.gov # NCT01531387).
TABLE 33.2 RECOMMENDED TRANSCRANIAL DOPPLER (TCD) SCREENING SCHEDULE IN CHILDREN WITH SICKLE CELL DISEASE | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
including ACS and pain episodes, and mortality.305, 306 A similar relationship was observed between asthma and the incidence of ACS in pediatric patients with Hb SC.307 Sickle cell patients with a history of asthma were four times more likely to develop ACS during a hospital admission for pain and had a substantially longer duration of hospitalization.308 Similarly, children with pulmonary function test (PFT)-documented lower airway obstruction had a greater risk for pain and ACS hospitalization (risk ratio 2.0, CI: 1.3 to 3.3).309 In Jamaican children with sickle cell disease, asthma and bronchiolar hyperreactivity were more common than in ethnic-matched controls and were associated with recurrent ACS.310 Much less information is available for adult patients, but a high prevalence of airway hyperresponsiveness to methacholine challenge was noted in those with a history of reactive airway disease.311 Hematologically, ACS is characterized by a sudden drop in Hb concentration and an increase in the number of platelets and leukocytes.312 Rib infarcts are a primary trigger of ACS when bone pain is followed by soft-tissue reaction, pleuritis, splinting, hypoventilation, atelectasis, and the typical radiologic picture.313 Incentive spirometry with the use of maximal inspirations every 2 hours has been shown to prevent ACS in patients with sickle cell disease who were hospitalized with chest or back pain.314 Lung crises may result from embolization of fat from infarcted bone marrow (pulmonary fat emboli)315 or deep-vein thrombi. Occlusion of major pulmonary vessels is a recognized cause of sudden death. Pulmonary fat emboli are found more commonly than previously appreciated when a diagnosis is sought by fat staining of pulmonary macrophages obtained by bronchoalveolar lavage.315 Fat emboli are associated with bone pain, chest pain, neurologic symptoms, acute decreases in Hb level and platelet count, and prolonged hospitalization. Secretory phospholipase A2 (sPLA2), an inflammatory mediator that liberates free fatty acids and lysophospholipids and may be responsible for acute lung injury, is markedly elevated in sickle cell patients 24 to 48 hours before the diagnosis of ACS in patients presenting with a pain event316, 317 and in most patients at the time of diagnosis of ACS.318 A clinical trial involved patients hospitalized for pain who developed fever and elevated sPLA2 and were randomized to receive transfusion or observation.158 Because of slow accrual, conclusions could not be reached regarding the effect of transfusion to pre-emptively avert an ACS event, but information regarding the sensitivity of this test was provided; a threshold level of sPLA2 ≥ 48 ng/ml offered 73% sensitivity and 71% specificity.
TABLE 33.3 CAUSES OF ACUTE CHEST SYNDROME | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
the gonadotropin-releasing hormone analog leuprolide acetate,346 low-dose antiandrogens,347 finasteride,348 and oral phosphodiesterase S inhibitor.349 Sildenafil is reported to improve priapism, but can trigger pain as a side effect.350 A polyethylene glycolmodified adenosine deaminase (PEG-ADA) has been used in transgenic sickle mice to reduce adenosine levels and promoted increased cavernosal relaxation, suggesting a possible new avenue of treatment for priapism.351 Despite either conservative or aggressive treatment, >25% of patients have some degree of impotence352 and may be candidates for a penile prosthesis after 6 to 12 months.353
common causes of hospitalization and previously was the most frequent cause of death, particularly during the first 3 years of life. S. pneumoniae is the usual infecting organism; the blood and spinal fluid are the major sites of infection.386, 387 Previously, the incidence of invasive infection with S. pneumoniae was ˜7/100 patient-years in children with SCA who were <5 years of age; this rate was 30 to 100 times that which would be expected in a healthy population of this age.387, 388 More than 70% of meningitis in children with SCA also resulted from S. pneumoniae.389 The mortality rate of pneumococcal sepsis was as high as 35%, but widespread improvement in parental education and aggressive management of the febrile child have greatly improved the likelihood of surviving a septic event.390 Furthermore, penicillin prophylaxis and pneumococcal vaccines have dramatically lowered the risk of invasive pneumococcal infection. Despite the dramatic decline in the rate of pneumococcal sepsis in recent decades (secondary to widespread use of pneumococcal immunization and penicillin prophylaxis), invasive pneumococcal infection still exists and may be life threatening.391, 392 A major threat to continued success in prevention and management of S. pneumoniae invasive infection has been the emergence of antibiotic-resistant pneumococcal organisms over the past two decades.391, 392
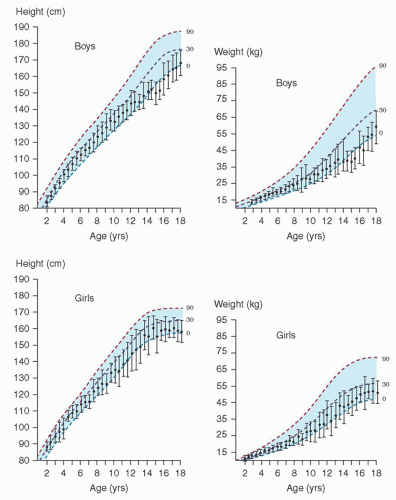 FIGURE 33.7. Height and weight (mean, ±1 standard deviation) of American boys and girls with sickle cell anemia (SCA) compared with National Center for Health Statistics growth percentiles. From Phebus CK, Gloninger MF, Maciak BJ. Growth patterns by age and sex in children with sickle cell disease. J Pediatr 1984;105:28, with permission. |
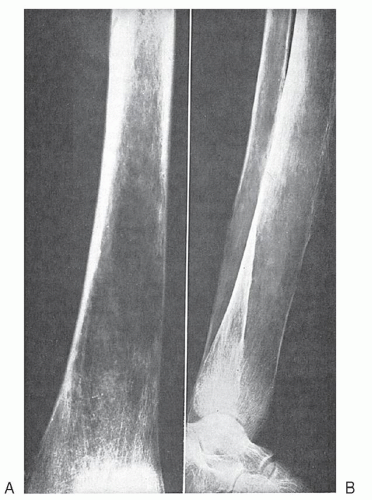 FIGURE 33.8. Sickle cell anemia. A: Femur. The cortex is thinned, and the normal bony architecture is disturbed. Adjoining small areas of translucency are areas of sclerosis. B: Tibia and fibula. Marked thinning of the cortex of the bones as well as periosteal reaction and disarrangement of the trabeculae. The latter changes and the extensive coarseness of the cortical layers suggest the bone is involved from within. |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


