Round Cell Lesions
Benign Lesions
Langerhans Cell Histiocytosis (Eosinophilic Granuloma)
Langerhans cell histiocytosis (LCH) is a lesion belonging to a group of disorders now classified by the World Health Organization (WHO) as histiocytic and dendritic cell disorders (39). The incidence in the United States is estimated at 0.05 to 0.5 per 100,000 children per year, with a 2:1 male predominance (20,45,49). This disorder represents less than 1% of all biopsy-proven primary bone lesions (16,20,52). Lichtenstein (46,47) proposed the name histiocytosis X for the three conditions: eosinophilic granuloma (condition with bone lesions only); Hand-Schüller-Christian disease or xanthomatosis (characterized by the triad of bone lesions, diabetes insipidus, and exophthalmos), now considered as multifocal unisystem disease; and Letterer-Siwe disease or nonlipid reticulosis (in which disseminated disease is marked by wasting, lymphadenopathy, hepatosplenomegaly, and anemia), now considered by the WHO as multifocal multisystem disease (75). His rationale was the postulation that these lesions, being of unknown cause, appeared to represent an increasing spectrum of severity in which the common denominator was the presence of the histiocyte (9,76). Schajowicz and Polak proposed the general term histiocytic granuloma for this group of disorders, which may appear without or with only isolated eosinophils or, more commonly, with many eosinophilic leukocytes, and may involve a single or several bones (68). Recently, histiocytosis X has been given the name of Langerhans cell histiocytosis (23,48,50) because it has been verified that the primary proliferative element in this disease is the Langerhans cell, a mononuclear cell of the dendritic type that is found in the epidermis but is derived from precursors in the bone marrow (60,61,67,70). LCH is characterized by its ultrastructural (Birbeck granules) and immunohistochemical (positivity for CD1a, S-100, CD68, and Langerin/CD207, the latter of which is involved in Birbeck granule formation) properties (6,28,62,71,72). Although its causes and pathogenesis remain unsettled, LCH is now considered a disorder of immune regulation rather than a neoplastic process (59,80). However, some laboratory studies have shown that certain lesions exhibit a clonal proliferation of cells, suggesting a neoplastic origin (77,79). In addition, differences between normal epidermal Langerhans cells
and lesional Langerhans cells have been described (44). Familial clustering and studies in twins with 80% and 33% concordance of the disease in monozygotic and dizygotic twins, respectively, point to a genetic component in LCH (4). Molecular genetic studies using comparative genomic hybridization (CGH) and loss of heterozygosity (LOH) experiments have revealed chromosomal alterations, with predominant losses affecting chromosomes 1p, 5p, 6q, 9, 16, 17, and 22q in CGH and highest LOH frequencies on 1p and 17, leading to the hypothesis that loss of tumor suppressor genes located on chromosome 1p may be involved in development and progression of the disease (57). Other studies have demonstrated alterations of the cell cycle in lesional Langerhans cells (22). Because lesions in LCH represent the cellular composition of an innate immune response, Nezelof and Basset (59) hypothesize that a block in the cross-talk between innate and adaptive immune responses may give rise to large numbers of inflammatory molecules and to the proliferation of Langerhans cells and macrophages. On the basis of these assumptions, Egeler et al. (22) proposed a unifying concept comprising a combination of oncogenesis (underlying genetic defect in replication pathways or DNA repair of Langerhans cells) and immune dysregulation (blockage of maturation of Langerhans cells in lesions).
and lesional Langerhans cells have been described (44). Familial clustering and studies in twins with 80% and 33% concordance of the disease in monozygotic and dizygotic twins, respectively, point to a genetic component in LCH (4). Molecular genetic studies using comparative genomic hybridization (CGH) and loss of heterozygosity (LOH) experiments have revealed chromosomal alterations, with predominant losses affecting chromosomes 1p, 5p, 6q, 9, 16, 17, and 22q in CGH and highest LOH frequencies on 1p and 17, leading to the hypothesis that loss of tumor suppressor genes located on chromosome 1p may be involved in development and progression of the disease (57). Other studies have demonstrated alterations of the cell cycle in lesional Langerhans cells (22). Because lesions in LCH represent the cellular composition of an innate immune response, Nezelof and Basset (59) hypothesize that a block in the cross-talk between innate and adaptive immune responses may give rise to large numbers of inflammatory molecules and to the proliferation of Langerhans cells and macrophages. On the basis of these assumptions, Egeler et al. (22) proposed a unifying concept comprising a combination of oncogenesis (underlying genetic defect in replication pathways or DNA repair of Langerhans cells) and immune dysregulation (blockage of maturation of Langerhans cells in lesions).
LCH exhibits a broad spectrum of clinical and radiologic abnormalities. It is characterized by an abnormal proliferation of Langerhans cells in various parts of the mononuclear system such as bone, lungs, central nervous system, skin, and lymph nodes.
Clinical Presentation
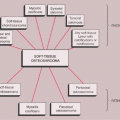
LCH may manifest as a solitary lesion or as multiple lesions (30,51). It occurs most often during childhood, with a peak incidence between the ages of 5 and 10 years (78). It is slightly more common in males (24). The most commonly affected sites are the skull (calvaria, skull base, and facial bones), the ribs, the pelvis, the spine, and the long bones (Fig. 5-1). Epiphyseal lesions are relatively rare (35,70). Clinical manifestations include local pain, tenderness, and swelling or a soft tissue mass adjacent to the site of the skeletal lesion (1). Fever, an elevated sedimentation rate, and leukocytosis may also be present (65). If a vertebra is affected, the patient may present with neurologic symptoms (16) resulting from a collapse of the vertebral body (31,41).
Imaging
Radiography remains the most effective modality for the diagnosis of LCH. In the mandible, radiolucent lesions and “floating teeth” secondary to the destruction of the alveolar bone are characteristic (Fig. 5-2). In the skull, a beveled lytic lesion is typical (Fig. 5-3) (25). In the center of the lytic lesion there is sometimes a small sclerotic focus referred to as a button sequestrum (11), “cockade” image (in European literature), or “bull’s eye” simulating an infectious process (16,46,65). In the spine, so-called vertebra plana (or coin-on-edge appearance) is a usual manifestation and results from a collapsed vertebral
body (10) (Fig. 5-4). This finding was for a long time mistakenly considered an osteochondrosis of the vertebra and was (and in the European literature still is) named Calvé disease after its proposer (10).
body (10) (Fig. 5-4). This finding was for a long time mistakenly considered an osteochondrosis of the vertebra and was (and in the European literature still is) named Calvé disease after its proposer (10).
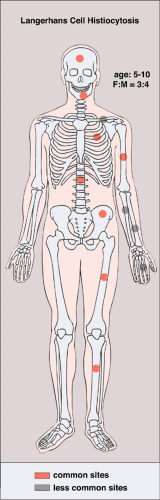 Figure 5-1 Langerhans cell histiocytosis: skeletal sites of predilection, peak age range, and male-to-female ratio. |
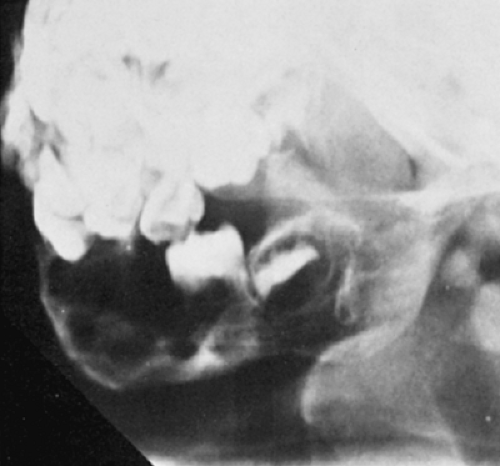 Figure 5-2 Langerhans cell histiocytosis. A 3-year-old girl with extensive skeletal involvement had in addition a large destructive lesion in the mandible. Note the characteristic appearance of a floating tooth, which results from destruction of supportive alveolar bone. |
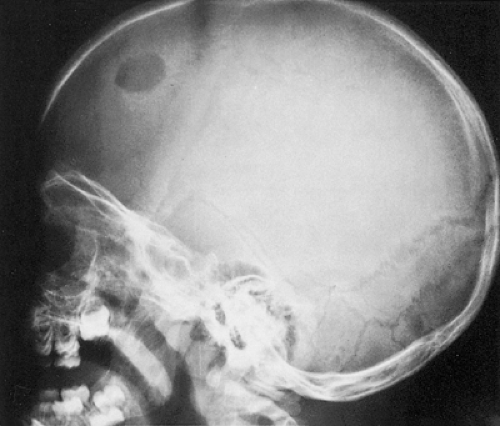 Figure 5-3 Langerhans cell histiocytosis. Lateral radiograph of the skull of a 2½-year-old boy with disseminated process shows an osteolytic lesion in the frontal bone with a sharply outlined margin, giving it a punched-out appearance. Uneven involvement of the inner and outer tables results in its beveled presentation. |
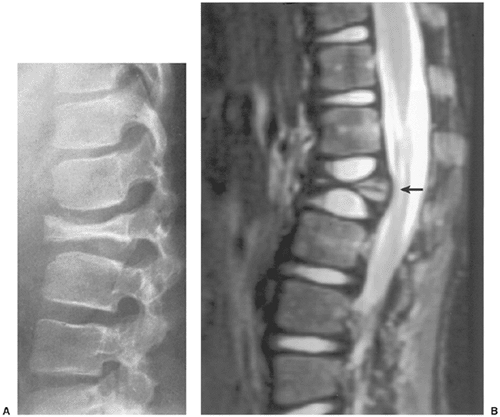 Figure 5-4 Langerhans cell histiocytosis. A: Vertebra plana represents collapse of vertebral body secondary to destruction of bone by granulomatous lesion. Note the preservation of the intervertebral disk space. B: In another patient, a sagittal T2-weighted magnetic resonance imaging (MRI) scan shows compression of the ventral aspect of the thecal sac by a fractured vertebral body (arrow). |
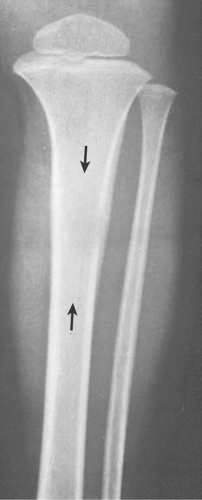 Figure 5-5 Langerhans cell histiocytosis. Anteroposterior radiograph of the lower leg of a 4-year-old boy demonstrates a lesion in the diaphysis of the left tibia (arrows) exhibiting a permeative type of bone destruction and a lamellated (onion-skin) type of periosteal reaction similar to that seen in osteomyelitis and Ewing sarcoma. |
In the long bones, LCH presents as a radiolucent destructive lesion, commonly associated with a lamellated periosteal reaction (27,29,67) (Fig. 5-5). This appearance may mimic that of a round cell malignant tumor, such as lymphoma or Ewing sarcoma. The lesion may have well-defined or poorly defined margins, with or without sclerotic borders (11). Occasionally, a cluster of partially overlapping lesions may be observed. In such instances, the superimposed areas of destruction have the appearance of a hole within a hole (55,56). A distinct and characteristic feature of many lesions is slanting or beveling of the edges (38). Sometimes the lesion is poorly demarcated and its indistinct borders gradually merge with the adjacent normal bone. The lesion may affect only the cortical surface of bone (21), although more commonly it is intramedullary, encroaching on the endocortex and causing scalloping of the endosteal surface (34) (Fig. 5-6). In later stages the lesion becomes more sclerotic, with dispersed radiolucencies (Fig. 5-7). The periosteal reaction is either absent or more defined and appears nonaggressive. Uhlinger (73) and later Mirra and Gold (52) have distinguished three phases in the evolution of LCH: incipient, midphase, and late phase. During the early phases the lesions tend to have aggressive patterns, with a lamellated periosteal reaction and poorly marginated or permeative lytic lesions. During the late phase the lesions tend to become more circumscribed (52).
The distribution of the lesion may be determined by radionuclide bone scan, which can aid in the detection of silent lesions and in differentiating LCH from Ewing
sarcoma, which rarely has multiple foci (43,69). The study has its limitations, however, because approximately 35% of lesions show a normal uptake of the radiopharmaceutical tracer (36). Bone destruction may sometimes result in inadequate residual bone to produce uptake, and areas of photon deficiency are exhibited (2). Recent reports indicate the usefulness of thallium-201 scintigraphy, which proved to be positive in a lesion that exhibited photon deficiency on technetium-99m methylene diphosphonate (99mTc-MDP) scintigraphy (26).
sarcoma, which rarely has multiple foci (43,69). The study has its limitations, however, because approximately 35% of lesions show a normal uptake of the radiopharmaceutical tracer (36). Bone destruction may sometimes result in inadequate residual bone to produce uptake, and areas of photon deficiency are exhibited (2). Recent reports indicate the usefulness of thallium-201 scintigraphy, which proved to be positive in a lesion that exhibited photon deficiency on technetium-99m methylene diphosphonate (99mTc-MDP) scintigraphy (26).
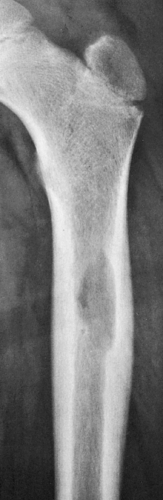 Figure 5-6 Langerhans cell histiocytosis. Anteroposterior radiograph of the proximal femur of a 3-year-old boy with a limp and tenderness localized to the upper thigh shows an osteolytic lesion without sclerotic changes, causing scalloping of the lateral endocortex. There is fusiform thickening of the cortex and a solid uninterrupted type of periosteal reaction. |
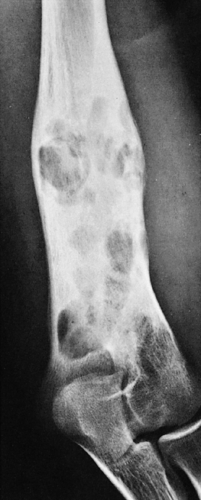 Figure 5-7 Langerhans cell histiocytosis. The healing stage of the lesion, as seen here in the distal humerus of a 16-year-old girl, exhibits predominantly sclerotic changes with interspersed radiolucent foci, thickening of the cortex, and a well-organized periosteal reaction. In this stage, the lesion mimics chronic osteomyelitis. |
Computed tomography (CT) may be useful if radiography inadequately defines the extent of the process (53), particularly in cases of spine and pelvic involvement (33). This modality effectively demonstrates periosteal reaction, beveled edges, and reactive sclerosis.
There have been isolated reports of the usefulness of magnetic resonance imaging (MRI) in evaluating this condition (5,40). The MRI appearance varies and appears to correlate with the radiographic appearance (11). The MRI manifestations of LCH during the earlier stages are nonspecific and may simulate an aggressive lesion, such as osteomyelitis or Ewing sarcoma, and occasionally benign tumors, such as osteoid osteoma or chondroblastoma (5). After gadolinium– diethylenetriamine-penta-acetic (DTPA) injection, the lesions show marked enhancement on T1-weighted images (19) (Fig. 5-8). Occasionally, MRI can demonstrate early bone marrow involvement in the absence of radiographic or scintigraphic abnormalities (45). In some studies, on T1-weighted sequences the lesions were isointense with adjacent structures (54,58). In the skull, lesions have been reported to show well-defined high-signal-intensity areas of marrow replacement on T2-weighted sequences. The most recent investigations have shown that the most common MRI appearance of LCH is that of a focal lesion, surrounded by an extensive, ill-defined signal from bone marrow and by soft tissue reaction with low signal intensity on T1-weighted images and high signal intensity on T2-weighted images, considered to represent bone marrow and soft tissue edema or the flare phenomenon (5,29) (Fig. 5-9).
Histopathology
Histologically, LCH consists of agglomerates of more or less densely arranged large rounded cells without cytoplasmic extensions, thus distinguishing them from epidermal Langerhans cells (Fig. 5-10). Electron microscopic observations proved that this pathognomonic cell was identical to the Langerhans cells seen in the skin (70,78). These cells are plump and pink, with a lightly granular cytoplasm and dendritic extensions. Their nuclei are translucent, ovoid, or kidney bean–shaped, have distinct nuclear membranes, and may exhibit longitudinal grooves. Langerhans cells can be specifically identified in electron microscopy by the presence of racquet-shaped cytoplasmic organelles known as Birbeck granules (6,70), also referred to as Langerhans cell granules or X granules (45) (see Fig. 1-43). In LCH, osteoclast-like giant cells are present, usually with five to seven nuclei (13). They may either represent osteoclasts, as indicated by their immunoprofile (positive for CD68, tartrate-resistant acid phosphatase, vitronectin receptor, cathepsin K, and metalloproteinase 9) or, because some giant cells also express CD1a, especially in nonbony lesions such as skin and lymph nodes, may be formed at a later stage when mononuclear bone marrow–based progenitor cells, capable of differentiating to osteoclasts or dendritic cells, have already switched to the dendritic pathway (3,66).
Langerhans cells express a variety of surface antigens. The IHC demonstration of CD1a, S-100 protein, and Langerin/CD207 and the electron microscopic (EM) identification of Birbeck granules are the most useful markers for Langerhans cells (8,20,32,62,74).
Zones of necrosis are common. In older or multiple lesions, lipid-bearing foam cells can also be observed (68) (Fig. 5-11). Special stains can reveal abundant droplets of sudanophilic fat peripherally or in the middle of the giant cell cytoplasm (so-called Touton cells) (68). The granulomatous lesions develop during the first (incipient) phase (52,73). During the second phase (midphase), more and more eosinophilic granulocytes
containing lobulated nuclei and coarse eosinophilic granules infiltrate the granuloma, finally forming dense agglomerates, the so-called eosinophilic pseudoabscesses (Fig. 5-12). Sparse infiltrates of lymphocytes and plasma cells may be found, as well as giant cells and foci of hemorrhage. The phagocytic capacity of mononuclear and multinucleated histiocytic cells can be demonstrated by their ingestion of erythrocytes, eosinophils, and hemosiderin granules (38). The cytoplasm of the phagocytic cells often exhibits double-refractile neutral fat deposition (38). The eosinophilic granulocytes then progressively disappear. Some of these features are displayed to better advantage with methylene blue and Giemsa preparations. In areas where the eosinophilic leukocytes are undergoing fragmentation, proteinaceous crystalline structures, the so-called Charcot-Leyden crystals, are demonstrable (68). In the third phase (late phase), fibroblasts, probably under the influence of transforming growth factor beta, a major mediator of fibrosis (12,17), produce collagen with concomitant reduction and final loss of Langerhans cells. The lesion then becomes indistinguishable from a focus of unspecific chronic fibroblastic osteomyelitis.
containing lobulated nuclei and coarse eosinophilic granules infiltrate the granuloma, finally forming dense agglomerates, the so-called eosinophilic pseudoabscesses (Fig. 5-12). Sparse infiltrates of lymphocytes and plasma cells may be found, as well as giant cells and foci of hemorrhage. The phagocytic capacity of mononuclear and multinucleated histiocytic cells can be demonstrated by their ingestion of erythrocytes, eosinophils, and hemosiderin granules (38). The cytoplasm of the phagocytic cells often exhibits double-refractile neutral fat deposition (38). The eosinophilic granulocytes then progressively disappear. Some of these features are displayed to better advantage with methylene blue and Giemsa preparations. In areas where the eosinophilic leukocytes are undergoing fragmentation, proteinaceous crystalline structures, the so-called Charcot-Leyden crystals, are demonstrable (68). In the third phase (late phase), fibroblasts, probably under the influence of transforming growth factor beta, a major mediator of fibrosis (12,17), produce collagen with concomitant reduction and final loss of Langerhans cells. The lesion then becomes indistinguishable from a focus of unspecific chronic fibroblastic osteomyelitis.
The so-called Langerhans cell sarcoma represents an exceedingly rare but very aggressive form of LCH with multiorgan involvement (75). It can arise de novo or may progress from typical LCH. The cells present with obvious malignant features such as enlarged nucleoli, unevenly distributed nuclear chromatin, and a high mitotic rate. By immunohistochemistry (IHC) they express CD1a and S-100 but sometimes only focally (74).
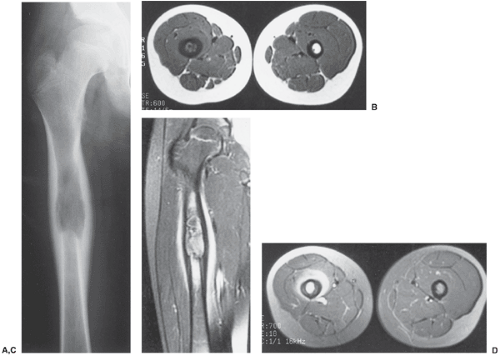 Figure 5-8 Langerhans cell histiocytosis: magnetic resonance imaging (MRI). A: Anteroposterior radiograph of the right femur of a 13-year-old boy shows a radiolucent lesion in the proximal femoral diaphysis exhibiting a lamellated periosteal reaction. B: Axial T1-weighted (SE, TR 600, TE 14) MRI demonstrates the lesion to be of low signal intensity. The cortex is markedly thickened. C: Coronal T1-weighted (SE, TR 500, TE 15) MRI obtained after intravenous injection of gadolinium shows marked enhancement of the lesion and the soft tissues adjacent to the thickened femoral cortex. D: Axial T1-weighted (SE, TR 700, TE 18) MRI obtained after intravenous injection of gadolinium shows enhancement of both granuloma and perilesional edema. |
Differential Diagnosis
Radiology
The differential diagnosis depends on the site and extent of bone involvement, the phase of the disease, and
the age of the patient (63,70). In a solitary lesion in long or flat bones, the main diagnostic considerations are Ewing sarcoma and osteomyelitis (see Fig. 5-5). Both lesions may exhibit moth-eaten or permeative types of bone destruction and lamellated, onion-skin periosteal reaction. If a solitary lesion of LCH in a long tubular bone does not develop a periosteal reaction, a differential diagnosis of a simple bone cyst must be considered. However, the characteristic clinical features of the former lesion should always be taken into consideration: the so-called tempo phenomenon (38) is a useful sign, i.e., progression and disappearance of the bony lesion is very rapid. The differential diagnosis of a solitary skull lesion in a child or young adult should include osteomyelitis, hemangioma, and fibrous dysplasia. In an elderly patient, the considerations are the lytic phase of Paget disease (osteoporosis circumscripta), myeloma, and a metastasis. The beveled edge or double-contoured appearance of the lesion is always highly suggestive of LCH.
the age of the patient (63,70). In a solitary lesion in long or flat bones, the main diagnostic considerations are Ewing sarcoma and osteomyelitis (see Fig. 5-5). Both lesions may exhibit moth-eaten or permeative types of bone destruction and lamellated, onion-skin periosteal reaction. If a solitary lesion of LCH in a long tubular bone does not develop a periosteal reaction, a differential diagnosis of a simple bone cyst must be considered. However, the characteristic clinical features of the former lesion should always be taken into consideration: the so-called tempo phenomenon (38) is a useful sign, i.e., progression and disappearance of the bony lesion is very rapid. The differential diagnosis of a solitary skull lesion in a child or young adult should include osteomyelitis, hemangioma, and fibrous dysplasia. In an elderly patient, the considerations are the lytic phase of Paget disease (osteoporosis circumscripta), myeloma, and a metastasis. The beveled edge or double-contoured appearance of the lesion is always highly suggestive of LCH.
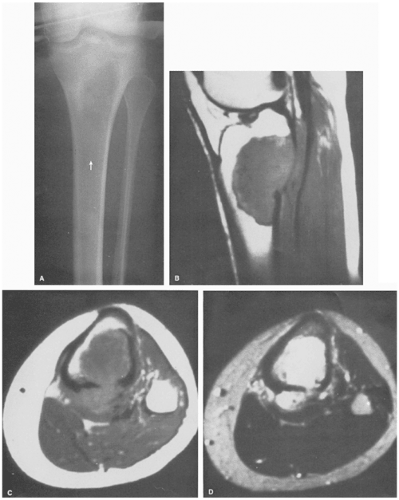 Figure 5-9 Langerhans cell histiocytosis: magnetic resonance imaging (MRI). A: Anteroposterior radiograph of the left tibia of a 20-year-old woman shows an ill-defined osteolytic lesion in the proximal part of the bone (arrow). B: T1-weighted sagittal MRI shows a lobulated lesion exhibiting a low signal intensity. C: T1-weighted axial MRI shows that the lesion penetrated the posterior cortex and extended into the soft tissues. D: T2-weighted axial MRI shows high signal intensity of the lesion in and outside the tibia. In addition, a high signal intensity soft tissue reaction surrounds the bone. (Reprinted with permission from Greenfield GB, Arrington JA. Imaging of bone tumors: a multimodality approach. Philadelphia: JB Lippincott, 1995:417.) |
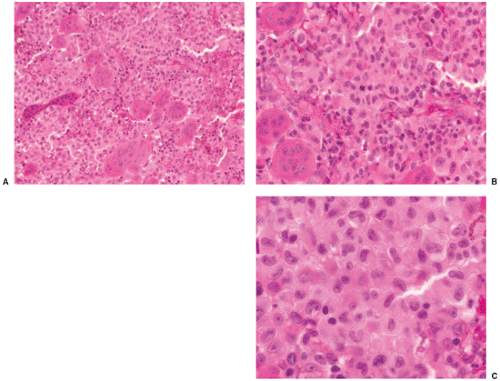 Figure 5-10 Histopathology of Langerhans cell histiocytosis. A: Densely arranged large Langerhans cells and rounded giant cells are infiltrated by numerous eosinophilic granulocytes (hematoxylin and eosin, original magnification ×100). B: At higher magnification large pale histiocytes (Langerhans cells) and numerous roundish giant cells containing up to ten nuclei (left) are clearly discernible. In addition, sparse lymphocytes and eosinophils are present (hematoxylin and eosin, original magnification ×400). C: At high power, the indentations of oval nuclei are clearly visible (hematoxylin and eosin, original magnification ×630). |
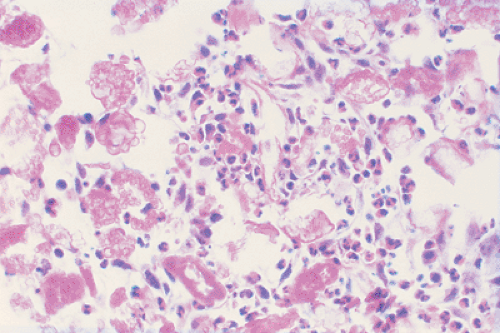 Figure 5-11 Histopathology of Langerhans cell histiocytosis. Mulberry-like foam cells (left and upper right), eosinophilic granulocytes, and hyaline bodies prevail in this field of view (hematoxylin and eosin, original magnification ×50). |
A lytic expansive lesion in the pelvis may mimic fibrous dysplasia, aneurysmal bone cyst, brown tumor of hyperparathyroidism, or hemophilic pseudotumor. Disseminated
osseous lesions in infants should be distinguished from infantile myofibromatosis (64), in adolescents and young adults must be distinguished from multifocal osteomyelitis, leukemia, lymphoma, cystic angiomatosis, fibrous dysplasia, brown tumors of hyperparathyroidism, and in older patients from metastatic disease and multiple myeloma.
osseous lesions in infants should be distinguished from infantile myofibromatosis (64), in adolescents and young adults must be distinguished from multifocal osteomyelitis, leukemia, lymphoma, cystic angiomatosis, fibrous dysplasia, brown tumors of hyperparathyroidism, and in older patients from metastatic disease and multiple myeloma.
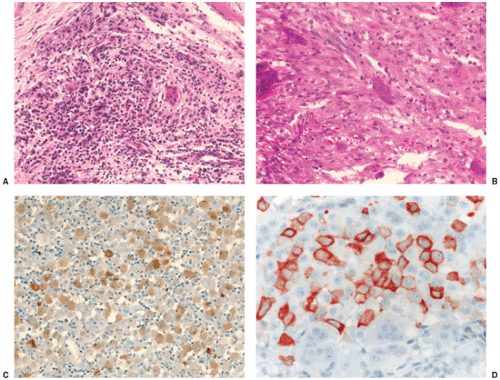 Figure 5-12 Histopathology of Langerhans cell histiocytosis. A: In a later stage of development, nonspecific inflammatory cells, particularly lymphocytes, predominate. Some giant cells are also present (hematoxylin and eosin, original magnification ×100). B: In another field, Langerhans cells exhibit a more spindle-shaped appearance. Scattered eosinophils and some giant cells are also present (hematoxylin and eosin, original magnification ×200). C: Tumor cells are positive for S-100 protein, both cytoplasmic and nuclear (biotin-streptavidin peroxidase, original magnification ×200). D: On higher magnification observe cytoplasmic and membranous positivity for CD1a in Langerhans cells (ABC peroxidase, original magnification ×400). |
Pathology
The major histologic differential diagnostic condition is infection because osteomyelitis is among the diseases capable of producing a polymorphous infiltrate of several inflammatory cell types. Moreover, there is an inflammatory reaction that simulates osteomyelitis, particularly during the midphase of LCH, and it is therefore necessary to correlate histologic findings with the radiographic presentation before rendering a diagnosis. Furthermore, when Langerhans cells and eosinophils have disappeared in the late phase, chronic productive osteomyelitis cannot be differentiated by histologic means.
Another diagnostic consideration should be Hodgkin lymphoma, which can present as a solitary osseous defect with a mixed inflammatory infiltrate. The identification of Reed-Sternberg cells and the absence of Langerhans cells in the latter serve as positive diagnostic clues. Furthermore, because the presentation of Hodgkin lymphoma as a primary disease in bone is exceedingly rare, clinical history is helpful (37,42).
During the involutional stage of LCH, the eosinophils markedly diminish in number and disappear altogether as the lesion progresses (matures). The large mononucleated histiocytes may become lipid-laden in this stage. The presence of those foci of foam cells may lead to the mistaken diagnosis of xanthoma of bone, juvenile
xanthogranuloma of bone, or Chester-Erdheim disease (7,14,15,18).
xanthogranuloma of bone, or Chester-Erdheim disease (7,14,15,18).
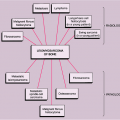
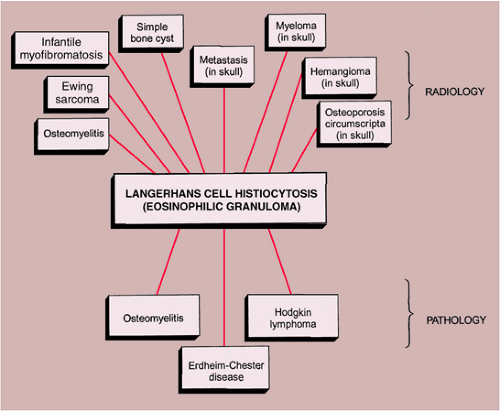 Figure 5-13 Radiologic and pathologic differential diagnosis of Langerhans cell histiocytosis. |
The radiologic and pathologic differential diagnosis of LCH is depicted in Figure 5-13.
Malignant Tumors
Round small cell malignancies of bone form a heterogeneous group of neoplasms, distinguished from most other malignancies of bone by the fact that the tumors form a pure cellular growth without production of a tumor matrix (90). The common denominator is an undifferentiated, round cell, basophilic, cytoplasm-poor, stroma-poor, highly cellular tumor (144). These malignancies include primary bone tumors such as Ewing sarcoma and primitive neuroectodermal tumor (PNET), lymphoma, mesenchymal chondrosarcoma, myeloma, metastatic neuroblastoma, and some other metastatic lesions (Table 5-1).
Ewing Sarcoma
Ewing sarcoma, along with Askin tumor and PNET, is now regarded as a single entity (Ewing sarcoma family of tumors) (113). These lesions are characterized by various degrees of neuroectodermal differentiation and by common histologic, immunohistochemical, and molecular properties (87,110,113,121,122,148).
Ewing sarcoma is the prototype of round, small cell malignancies of bone. It is the sixth most common malignant tumor, comprising approximately 11% (138) to 12% (127) of all malignant tumors of bone, with a rather uniform histologic appearance. Conventional Ewing sarcoma is composed of densely packed small cells with round nuclei but without distinct cytoplasmic outlines (138). It exhibits no microscopic evidence of matrix production (97). Occasionally, similar tumors can arise as primary neoplasms of soft tissue. These are referred to as extraskeletal or soft tissue Ewing sarcomas (95). Even less commonly, Ewing sarcoma may exhibit periosteal location (82). There is also a rare form of Ewing sarcoma in which the cells are larger and more pleomorphic than those of conventional Ewing tumor. This form has been referred to as atypical Ewing sarcoma or large cell Ewing sarcoma (129). The clinical behavior and prognosis of large cell Ewing sarcoma appear similar to those of conventional tumor. Furthermore, single cases of so-called adamantinoma-like Ewing sarcomas have been described. These sarcomas present with tumor cell nests or anastomosing cords of tumor cells and strong cytokeratin positivity but reveal the translocation typical of Ewing sarcomas (see later), which is not present in adamantinoma of long bones (85,106). Very rarely, Ewing sarcoma may be multifocal (88).
All tumors of the Ewing family are characterized by recurrent chromosomal translocations involving chromosomes 11 and 22 [t(11;22)(q24;q12)] or chromosomes 21 and 22 [t(21;22)(q22;q12)] in about 85% or 15% of cases, respectively. In less than 1% of cases, translocations involving chromosomes 22 and chromosomes 2, 7, or 17 and inversions of chromosome 22 (i.e., breakage of a single chromosome with consecutive reversed insertion) have been described. These
transcripts give rise to fusions between EWS (for Ewing sarcoma) gene on chromosome 22q12 and members of the ETS family of transcription factors (ETS is derived from a sequence that was detected in an avian erythroblastosis virus, E26, where it formed a transforming gene; it was therefore called E26 transformation specific sequence or ETS) like Fli1 (for Friend leukemia virus integration 1; present in about 85%), ERG (early response gene; present in about 15%), ETV1 (or ETS translocation variant-1), E1A-F (for E1A factor, a binding protein of the adenovirus E1A gene), and FEV (for fifth Ewing variant), or ZSG (for zinc finger sarcoma gene), a transcription activator (136,148). As a result, chimeric proteins are expressed, which are believed to act as aberrant transcription factors that may upregulate or downregulate Ewing sarcoma target genes. The gene for the transforming growth factor (TGF) beta II receptor, a putative tumor suppressor gene, has been identified as such a target that is downregulated by the EWS/Fli1 transcript (105). Recently, additional putative target genes of EWS/Fli1 have been recovered from DNA of Ewing sarcomas (140).
transcripts give rise to fusions between EWS (for Ewing sarcoma) gene on chromosome 22q12 and members of the ETS family of transcription factors (ETS is derived from a sequence that was detected in an avian erythroblastosis virus, E26, where it formed a transforming gene; it was therefore called E26 transformation specific sequence or ETS) like Fli1 (for Friend leukemia virus integration 1; present in about 85%), ERG (early response gene; present in about 15%), ETV1 (or ETS translocation variant-1), E1A-F (for E1A factor, a binding protein of the adenovirus E1A gene), and FEV (for fifth Ewing variant), or ZSG (for zinc finger sarcoma gene), a transcription activator (136,148). As a result, chimeric proteins are expressed, which are believed to act as aberrant transcription factors that may upregulate or downregulate Ewing sarcoma target genes. The gene for the transforming growth factor (TGF) beta II receptor, a putative tumor suppressor gene, has been identified as such a target that is downregulated by the EWS/Fli1 transcript (105). Recently, additional putative target genes of EWS/Fli1 have been recovered from DNA of Ewing sarcomas (140).
Table 5-1 Small Round Cell Tumors of Bone | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
In about 20% of cases of Ewing sarcoma, the second most common genetic alteration is the inactivation of the gene p16 or INK4A, which is involved in cell-cycle control and is responsible for preventing cells from entering the S-phase by suppressing the activity of the retinoblastoma protein (116). Wei et al. (152) suggested that p16 deletions represent a significant negative predictive factor in Ewing sarcoma. This has recently been confirmed by the same group and has been shown for mutations of TP53, which are present in about 10% of cases (108). In contrast, the most common type of EWS/Fli1 fusion (type 1 fusion between EWS exon 7 and Fli1 exon 6) is associated with a better prognosis (148).
Clinical Presentation
Ewing sarcoma occurs primarily in young people, most commonly in the second decade. Approximately 90% of cases present before the age of 20 years, with a median age of about 13 years (130). The tumor has a decisively male predominance (3:2), and only exceptionally occurs in black individuals (138). Clinically, Ewing sarcoma may present as a localized, painful mass or with systemic
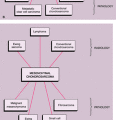
symptoms such as fever, malaise, weight loss, leukocytosis, and increased erythrocyte sedimentation rate (115), a constellation of findings that may lead to an erroneous diagnosis of osteomyelitis (99). It has been speculated that the presence of systemic symptoms indicates dissemination of the tumor, with a consequently worsened prognosis (83). Diaphyses of the long bones (predominantly femur, tibia, and humerus), the ribs, and the flat bones, such as the scapula and the pelvis, are preferred sites (90,118) (Fig. 5-14). Although metaphyses of the long bones occasionally may be affected (135), an epiphyseal location is rare (2%) (125,138). The small bones of the hands and feet can also be affected (81). According to Ilaslan et al. (109), 9.8% of all cases of Ewing sarcoma has a primary vertebral origin. Pathologic fractures are uncommon (97). Occasionally, Ewing sarcoma may metastasize to the other bones.
symptoms such as fever, malaise, weight loss, leukocytosis, and increased erythrocyte sedimentation rate (115), a constellation of findings that may lead to an erroneous diagnosis of osteomyelitis (99). It has been speculated that the presence of systemic symptoms indicates dissemination of the tumor, with a consequently worsened prognosis (83). Diaphyses of the long bones (predominantly femur, tibia, and humerus), the ribs, and the flat bones, such as the scapula and the pelvis, are preferred sites (90,118) (Fig. 5-14). Although metaphyses of the long bones occasionally may be affected (135), an epiphyseal location is rare (2%) (125,138). The small bones of the hands and feet can also be affected (81). According to Ilaslan et al. (109), 9.8% of all cases of Ewing sarcoma has a primary vertebral origin. Pathologic fractures are uncommon (97). Occasionally, Ewing sarcoma may metastasize to the other bones.
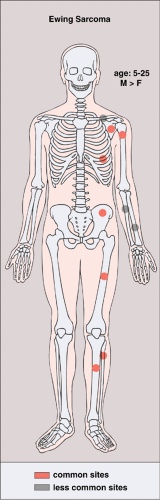 Figure 5-14 Ewing sarcoma: skeletal sites of predilection, peak age range, and male-to-female ratio. |
Imaging
In most cases, the radiographic presentation of Ewing sarcoma is quite characteristic. It consists of an ill-defined lesion with a permeative or moth-eaten pattern of bone destruction associated with a lamellated periosteal new bone formation that has an onion skin (or “onion peel”) appearance, or, less commonly, a “sunburst” (“trimmed whiskers” effect) configuration (101), and a large soft tissue mass (Fig. 5-15). Ewing sarcoma occasionally presents as a large area of geographic bone destruction, with or without matrix mineralization, simulating any other type of bone sarcoma. At times the lesion in the bone is almost imperceptible, and the only prominent finding is a soft tissue mass (Fig. 5-16). The lesion sometimes creates a typical “saucerization” of the cortex, a feature once believed to be virtually pathognomonic for this tumor (Fig. 5-17). The saucerization may be related to the combination of destruction of the periosteal surface of bone by the tumor and an associated extrinsic pressure effect of the large soft tissue mass (132). This sign has recently been reported in other tumors and even in osteomyelitis. However, the presence of saucerization associated with a permeative lesion and a soft tissue mass favors the diagnosis of Ewing sarcoma. When the lesion exhibits a permeative appearance, it may be indistinguishable from osteomyelitis (Fig. 5-18). Because Ewing sarcoma does not produce a matrix material, radiography reveals a lack of mineralization in the substance of the tumor. However, because there is often an abundant periosteal new bone formation, particularly in the flat bones, this appearance may mimic osteosarcoma, and differentiation between the two tumor types may be difficult (Fig. 5-19). Approximately one third of the cases affecting flat bones demonstrate a diffuse sclerosis. Unlike osteosarcoma, this sclerosis represents not a tumor bone but rather reactive bone to the tumor cell infiltration (139).
On radionuclide bone scan, Ewing sarcoma shows a very intense increase of technetium-99m methylene diphosphonate (99mTc-MDP) accumulation (128). Gallium-67-citrate (67Ga-citrate) more readily identifies soft tissue tumor extension (94,96). Although scintigraphic findings are nonspecific, this technique
provides reliable information concerning the presence of skeletal metastases (112,127).
provides reliable information concerning the presence of skeletal metastases (112,127).
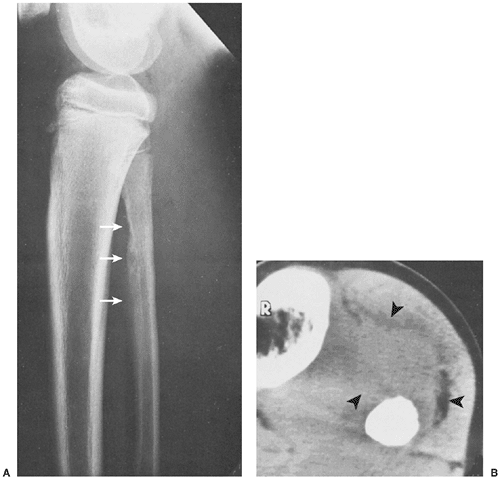 Figure 5-15 Ewing sarcoma. A: Lateral radiograph of a 12-year-old boy shows the typical appearance of this malignancy in the diaphysis of the fibula (arrows). The poorly defined lesion exhibits a permeative bone destruction associated with an aggressive periosteal reaction. B: CT section through the tumor demonstrates a large soft tissue mass (arrowheads), not well depicted on the routine study. Note the complete obliteration of the marrow cavity by tumor. |
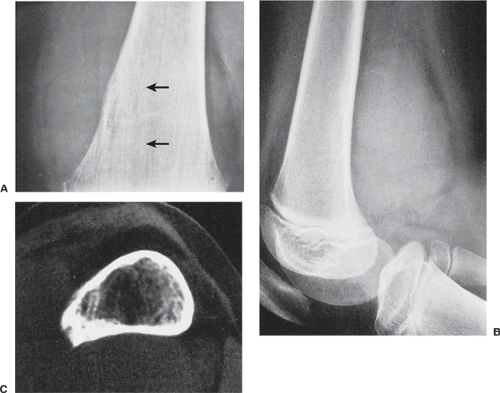 Figure 5-16 Ewing sarcoma. A: Bone destruction (arrows) is almost imperceptible on this magnification study of the distal femoral diaphysis of a 10-year-old girl. B: Lateral radiograph of the distal femur, however, shows a large soft tissue mass adjacent to the posterior cortex. C: Computed tomography using a bone window demonstrates destruction of the medullary portion of the bone and invasion of the cortex. |
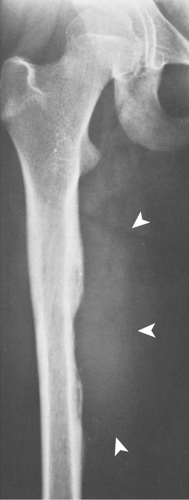 Figure 5-17 Ewing sarcoma. Anteroposterior radiograph of the right femur of a 12-year-old girl shows saucerization of the medial cortex of the diaphysis, often seen in Ewing sarcoma. Note a large soft tissue mass (arrowheads). |
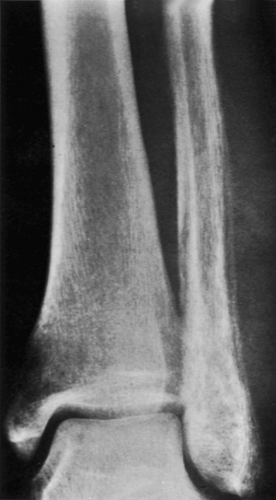 Figure 5-18 Ewing sarcoma resembling osteomyelitis. A 24-year-old man presented with pain and swelling of the left ankle for 8 weeks; he also had a fever. Anteroposterior radiograph of the ankle demonstrates a lesion of the distal fibula exhibiting a permeative type of bone destruction and a lamellated periosteal reaction. The appearance is that of infection (osteomyelitis), but biopsy revealed Ewing sarcoma. |
CT reveals the pattern of bone destruction, and attenuation values (Hounsfield units) provide information about the medullary extension (94). In addition, CT may help to delineate extraosseous involvement (149) (see Fig. 5-15B).
MRI is essential for definite demonstration of the extent of intraosseous and extraosseous involvement by this tumor (84,150) (Fig. 5-20). In particular, MRI may effectively reveal extension through the epiphyseal plate (100). T1-weighted images show intermediate to low signal intensity, which becomes bright on T2 weighting. Hypocellular regions and areas of necrosis are of lesser intensity (94). Imaging after injection of gadolinium (Gd-DTPA) reveals signal enhancement of the tumor on T1-weighted sequences (100). Enhancement occurs only in the cellular areas, allowing differentiation of the tumor from the peritumoral edema.
Histopathology
The gross appearance of Ewing sarcoma may be misleading (155). Because the lesion does not produce any matrix, its appearance can range from a soft and fleshy but solid mass to an almost liquid consistency. When a tumor of the latter consistency is cut into at the time of surgery, it may actually run like pus. This
can lead to a mistaken diagnosis of osteomyelitis, and the material may be sent for microbiologic culture rather than histopathologic examination, thus causing a delay in diagnosis (83).
can lead to a mistaken diagnosis of osteomyelitis, and the material may be sent for microbiologic culture rather than histopathologic examination, thus causing a delay in diagnosis (83).
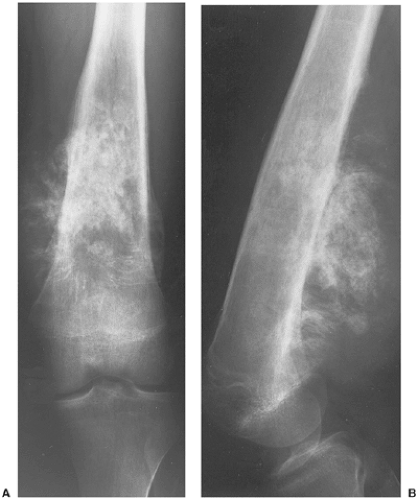 Figure 5-19 Ewing sarcoma resembling osteosarcoma. Anteroposterior (A) and lateral (B) radiographs of the left femur of a 17-year-old boy, displaying a significant sclerosis of the tumor that was originally interpreted as osteosarcoma. |
The microscopic appearance of Ewing sarcoma is typical. At lower magnification, the tumor consists of small, uniform-sized cells characterized by an almost clear to light eosinophilic cytoplasm and nuclei that are round and slightly hyperchromatic. Prominent septa composed of fibrous connective tissue divide the tumor tissue into strands or lobules (138) (Fig. 5-21A). Because the cell borders are indistinct, it sometimes appears as if the tumor is composed of many nuclei floating in a sea of cytoplasm rather than being separated into discrete cells. The nucleoli are usually inconspicuous. Areas of necrosis and hemorrhage are often observed (120). At higher magnification, the nuclei appear round and uniform (Fig. 5-21B–D). Sometimes the cytoplasm contains a moderate amount of glycogen that can be demonstrated by periodic acid–Schiff (PAS) staining (137) (Fig. 5-22) or, more specifically, with Best carmine stain with and without diastase digestion prior to staining, which will remove glycogen. Mitotic figures are uncommon in classical Ewing sarcoma (138). Although no extracellular matrix is found between the tumor cells, and reticulin fibers are almost always absent (Fig. 5-22C), reactive new bone formation may be observed, especially at the periphery. This reactive bone is sometimes difficult to distinguish from tumor-bone formation, and small cell osteosarcoma may therefore be erroneously diagnosed.
In extremely rare cases, Ewing sarcoma exhibits an epithelial differentiation similar to adamantinoma of the long bones (93,103,124,126,141,151). Other primary tumors of bone known to have epithelial features are adamantinoma of long bones (89), malignant fibrous histiocytoma (153), and osteosarcoma (91,117).
Electron microscopy of this lesion reveals highly characteristic ultrastructural features (133,142). The predominant or principal cell type is polygonal. The cytoplasm exhibits few organelles, with sparse rough endoplasm reticulum, and few mitochondria. However, a conspicuous Golgi complex is often present. The nuclei of these cells are round or oval. Typically, the chromatin is fine and evenly distributed, and there are single or multiple small nucleoli. The cytoplasm may contain many glycogen granules. In addition to
the principal cell, there may be a small number of so-called dark cells (reflecting a condensed chromatin) (119,138). It is still undetermined whether this cell is a precursor or, more likely, a terminal form of the principal cell.
the principal cell, there may be a small number of so-called dark cells (reflecting a condensed chromatin) (119,138). It is still undetermined whether this cell is a precursor or, more likely, a terminal form of the principal cell.
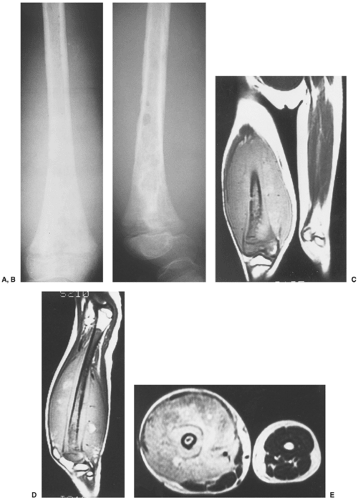 Figure 5-20 Ewing sarcoma: magnetic resonance imaging (MRI). Anteroposterior (A) and lateral (B) radiographs of the right distal femur of a 7-year-old girl show permeative type of bone destruction in the metaphysis and diaphysis associated with a large soft tissue mass. Coronal (C) and sagittal (D) T1-weighted (SE, TR 750, TE 20) MR images demonstrate the intraosseous and extraosseous extent of the tumor. E: Axial T2-weighted (SE, TR 2000, TE 80) MRI shows heterogeneous but mostly high signal intensity of the soft tissue mass. |
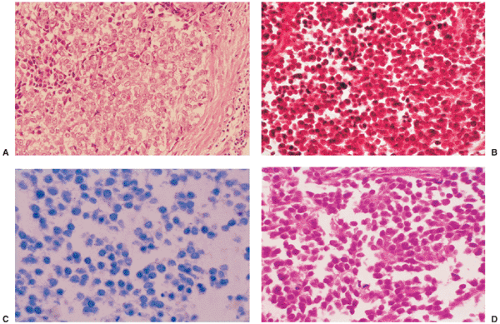 Figure 5-21 Histopathology of Ewing sarcoma. A: Densely packed small cells with round nuclei and indistinct cell borders are characteristic for this tumor. To the right a prominent septum composed of fibrous connective tissue is visible (hematoxylin and eosin, original magnification ×50). B: At high magnification the uniform aspect of the round nuclei without clear cytoplasmic outlines is striking (hematoxylin and eosin, original magnification ×100). C: With Giemsa staining the cytoplasm is more distinct. The nuclei are darkly and homogeneously stained (Giemsa, original magnification ×100). D: With alcohol fixation, in this case there was better preservation of cell details (hematoxylin and eosin, original magnification ×100). |
IHC reveals that almost all Ewing family tumors exhibit a positive membranous or cytoplasmic reaction for CD99 and vimentin, respectively (102) (Fig. 5-23). Because CD99 positivity [a transmembrane glycoprotein involved in cell adhesion, apoptosis and differentiation, encoded by the Mic2 gene which is located on both x and y chromosomes as a pseudoautosomal gene at the end of their short arms (114)] is not specific for Ewing tumors, additional markers covering the spectrum of “small round blue-cell” tumors must be applied (see later). To diagnose a given tumor as PNET, more than one finding of neural differentiation must be present, i.e., Homer-Wright rosettes by light microscopy or positive immunoreactions for two neural markers [neuron-specific enolase (NSE), synaptophysin, neurofilament, S-100, CD57, protein gene product 9.5 (PGP9.5)], at least in some tumor cells (Fig. 5-24), or neurosecretory granules within cytoplasmic processes must be demonstrated by ultrastructural analysis (131).
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


