Cartilage (Chondrogenic) Lesions
Diagnosis of a bone lesion as originating from cartilage is usually a simple task for the radiologist. The lesion’s radiolucent matrix, scalloped margin, and annular, punctate, or comma-shaped calcifications usually suffice to establish its chondrogenic nature (Fig. 3-1). However, whether a cartilage tumor is benign or malignant is sometimes extremely difficult for the radiologist and the pathologist to determine (33,49,62,70).
All cartilage tumors, whether benign or malignant, exhibit a positive reaction for S-100 protein (54). This is sometimes diagnostically useful, as most other bone tumors do not display this reaction. Histologically, cartilage tumors are usually recognized by the features of their intercellular matrix, which has a uniformly translucent appearance and contains less collagen compared with the intercellular matrix of osteoblastic tumors, such as osteoblastoma and osteosarcoma. The tumor cells themselves are usually located in rounded spaces (lacunae), as in normal cartilage. In benign cartilage tumors, such as enchondroma, the tissue is sparsely cellular (Fig. 3-2). The cells, typically with small, dark-staining nuclei, fail to show the cytologic features that are characteristic of chondrosarcoma, such as large nuclei; pleomorphism; large, swollen chondroblastic and double nuclei; and mitoses. The tumor tissue is usually avascular, and areas of degenerate and calcified matrix are common. Calcification is sometimes followed by vascularization and replacement by bone, as in normal endochondral ossification. Because of their slow growth, enchondromas erode and expand the overlying cortical bone (Fig. 3-3) but do not invade it, unlike chondrosarcomas. Nevertheless, some slow-growing, low-grade chondrosarcomas
can masquerade as enchondromas, and some aggressive-looking benign cartilage lesions may be mistakenly diagnosed as chondrosarcomas. Moreover, it is important to be aware of the capricious biological behavior displayed by cartilage tumors. Some benign-appearing lesions of cartilage may behave in an aggressive or malignant manner, whereas tumors that present an ominous appearance on radiologic or histologic examination may show limited progression.
can masquerade as enchondromas, and some aggressive-looking benign cartilage lesions may be mistakenly diagnosed as chondrosarcomas. Moreover, it is important to be aware of the capricious biological behavior displayed by cartilage tumors. Some benign-appearing lesions of cartilage may behave in an aggressive or malignant manner, whereas tumors that present an ominous appearance on radiologic or histologic examination may show limited progression.
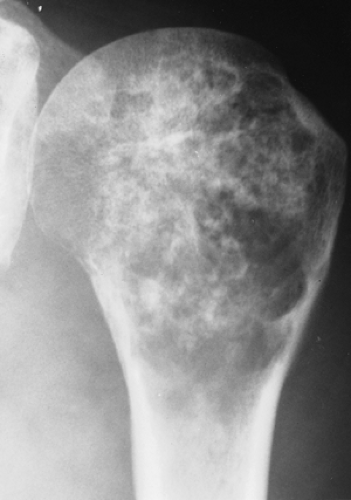 Figure 3-1 Cartilage-forming tumor. The radiologic characteristics of a radiolucent matrix, scalloped margin, and annular and comma-shaped calcifications establish the chondrogenic nature of this lesion (in this example—chondrosarcoma). |
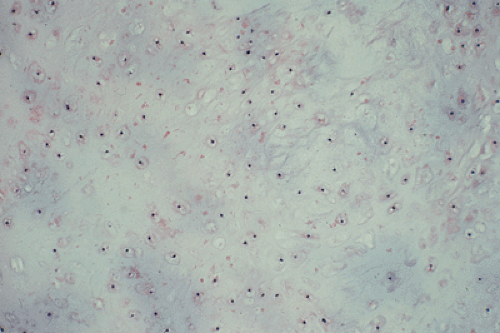 Figure 3-2 Histopathology of a benign cartilage lesion. Sparsely cellular tissue and uniform-sized chondrocytes located in rounded lacunae are characteristic features of enchondroma (hematoxylin and eosin, original magnification ×235). |
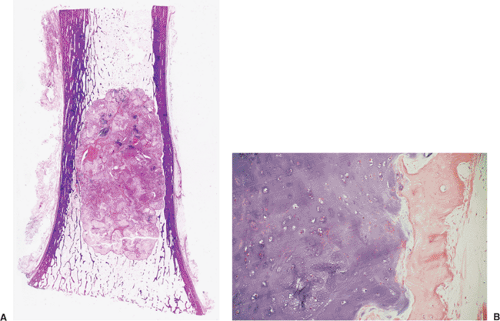 Figure 3-3 Histopathology of enchondroma. A: Whole-mount section (hematoxylin and eosin) shows the tumor eroding the cortex (endosteal scalloping). B: On higher magnification there is no evidence of invasion (hematoxylin and eosin, original magnification ×183). |
Clinically, malignancy is suggested by a variety of signs, including development of pain in a previously asymptomatic lesion (e.g., malignant transformation of enchondroma to chondrosarcoma), development of swelling or a mass at the site of a lesion, or rapid growth of a lesion (e.g., malignant transformation of osteochondroma to chondrosarcoma). Radiologically, the presence of a periosteal reaction, destruction of the cortex, and a soft tissue mass are almost pathognomonic features of malignancy (25). The histopathologic features of a malignant cartilage tumor include the presence of hypercellular and pleomorphic tumor tissue, appreciable numbers of plump cells with large or double nuclei, invasion (permeation) of bone trabeculae, and infiltration of Haversian systems and bone marrow (Fig. 3-4). However, these obvious features of malignancy may be not so prominent in biopsy specimen, or only a few areas in such a specimen may reveal the diagnostic features of malignancy. As Dahlin and Unni (13) pointed out, in the pathologic evaluation it is necessary to examine many fields in these tumors to be certain that there is sufficient evidence for a diagnosis of malignancy because evidence that a tumor is a chondrosarcoma rather than a benign enchondroma is frequently found in isolated areas within the mass. Therefore, it is of paramount importance to obtain biopsy specimens from several areas of a tumor. Close cooperation among the clinician, the radiologist, and the pathologist in reviewing the history, the radiologic examination, and the biopsy specimens is crucial to reaching a conclusion on the exact nature of a cartilage lesion.
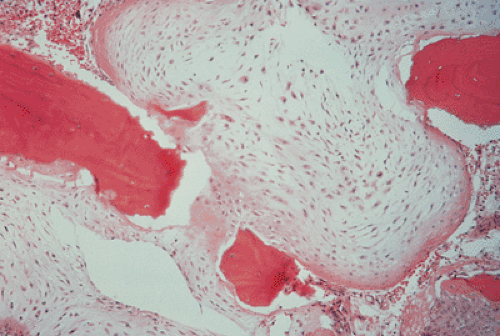 Figure 3-4 Histopathology of chondrosarcoma. Note invasion of trabeculae by markedly cellular tissue of chondrosarcoma (hematoxylin and eosin, original magnification ×25). (Courtesy Dr. M. J. Klein, Birmingham, Alabama.) |
From the radiologic standpoint, the differential diagnosis of cartilage lesions should focus on the following three points:
Distinguishing a benign cartilage tumor from the noncartilaginous lesions (e.g., calcifying enchondroma from bone infarct; enchondroma located near the articular end of bone from giant cell tumor, osteoarthritic geode, or intraosseous ganglion; or chondromyxoid fibroma from aneurysmal bone cyst).
Distinguishing a benign cartilage tumor from a malignant one (e.g., enchondroma from low-grade chondrosarcoma; osteochondroma from exostotic chondrosarcoma; periosteal chondroma from periosteal chondrosarcoma; or synovial chondromatosis from synovial chondrosarcoma).
Distinguishing a malignant cartilage tumor from a noncartilaginous lesion [e.g., chondrosarcoma at the articular end of bone from giant cell tumor or from malignant fibrous histiocytoma (MFH); or periosteal chondrosarcoma from periosteal osteosarcoma].
From the histopathologic standpoint, because the microscopic features of cartilage lesions are obvious and characteristic, focus should be directed to the following:
Recognition of the varieties of benign cartilage tumors (e.g., enchondroma, periosteal chondroma, chondroblastoma, chondromyxoid fibroma) and various subtypes of chondrosarcoma (e.g., clear cell, mesenchymal, or periosteal).
Recognition of benign versus malignant tumor (e.g., enchondroma vs. well-differentiated chondrosarcoma; chondromyxoid fibroma vs. myxoid chondrosarcoma; or chondroblastoma vs. mesenchymal chondrosarcoma).
Differentiation of chondrosarcoma from other malignant tumors that contain cartilage (e.g., from chondroblastic osteosarcoma or periosteal osteosarcoma).
Benign Lesions
Enchondroma
Enchondroma is the second most common benign tumor of bone and is characterized by the formation of mature hyaline cartilage. It constitutes about 10% of all benign bone tumors and represents the most common tumor of the phalanges of the hand. Several theories have been postulated regarding its etiology. Among these is that the enchondroma develops from abnormal zones of dysplastic chondrocytes in the growth plate. These abnormal foci fail to undergo normal endochondral ossification; instead, they are deposited within the metaphysis and, as the bone grows, are displaced into the diaphysis (49). Customarily, a lesion located centrally in the bone is called an enchondroma, whereas one located outside the cortex is called a chondroma (periosteal or juxtacortical). Separate groups
consist of lesions (a) located in the soft tissues and (b) located in the joint, bursa, or in the tendon sheath. The former are called soft tissue chondromas and the latter synovial chondromatosis (69).
consist of lesions (a) located in the soft tissues and (b) located in the joint, bursa, or in the tendon sheath. The former are called soft tissue chondromas and the latter synovial chondromatosis (69).
Clinical Presentation
Although the tumor may occur at any age, most cases arise between the second and fourth decades; there is no sex predilection. The short, tubular bones of the hand and foot (phalanges, metacarpals, and metatarsals) are preferred sites, followed in frequency by the femur, humerus, tibia, and ribs (Fig. 3-5). The lesion is often asymptomatic and usually is discovered incidentally due to a pathologic fracture through the tumor.
The single most important complication of the lesion is malignant transformation to chondrosarcoma. In a solitary enchondroma this occurs predominantly in a long or flat bone and almost never in a short tubular bone. An important clinical sign is development of pain at the site of the lesion in a previously asymptomatic patient and in the absence of a fracture.
Imaging
Radiography is usually sufficient to demonstrate the lesion and to establish its chondroid character. In short, tubular bones, the lesion is often entirely radiolucent (Fig. 3-6), but in long bones it may display visible calcifications (24). This flocculent mineralization usually assumes the form of dots, rings, and arcs, giving a “popcorn” appearance (Fig. 3-7). If the calcifications are extensive, the lesion is called a calcifying enchondroma (Fig. 3-8). The cortex is usually thinned and expanded in a symmetric, fusiform fashion. The often cloudy or hazy appearance of a lesion provides a clue to its chondroid composition (18). The lesion can also be recognized by scalloped inner cortical margins, reflecting the lobular growth pattern of cartilage (Fig. 3-9). Computed tomography (CT) and magnetic resonance imaging (MRI) may further delineate the tumor and more precisely localize it in the bone (Fig. 3-10). Because in long bones enchondroma may present as a radiolucent lesion with poorly defined margins, particularly in areas where trabeculae are sparse or absent (diaphysis), MRI may be extremely helpful in revealing the exact size and extent of the tumor (Fig. 3-11) (74). Like most cartilaginous and fibrous lesions, enchondroma exhibits low to intermediate signal intensity on T1-weighted images and high signal intensity on T2-weighted sequences. On gradient echo sequences the lesion may appear less bright than on conventional T2-weighted images (see Fig. 3-11E). The calcifications image either as signal void or as low-signal-intensity foci. After administration of gadolinium, enchondroma demonstrates a typical pattern of signal enhancement, which is helpful in distinguishing this lesion from noncartilaginous tumors (52,218).
Histopathology
On histologic examination, enchondroma consists of lobules of hyaline cartilage of variable cellularity and can be recognized by the features of its intercellular matrix, which has a uniformly translucent appearance
and contains relatively little collagen (Fig. 3-12A). The lesion may slightly erode the endosteal surface of cortical bone; however, no invasion of Haversian systems is present. The tumor cells are located in lacunae. The tissue is sparsely cellular and the cells contain small, dark-staining nuclei (Fig. 3-12B). Binucleated cells and small nucleoli are sometimes present (51) (Fig. 3-12C). Calcifications are common and correspond to matrix calcifications or actual endochondral ossifications at the periphery of cartilage lobules (Fig. 3-13). Enchondromas located in the short tubular bones of hands and feet may exhibit increased cellularity, plump nuclei, and occasionally even binucleated cells.
and contains relatively little collagen (Fig. 3-12A). The lesion may slightly erode the endosteal surface of cortical bone; however, no invasion of Haversian systems is present. The tumor cells are located in lacunae. The tissue is sparsely cellular and the cells contain small, dark-staining nuclei (Fig. 3-12B). Binucleated cells and small nucleoli are sometimes present (51) (Fig. 3-12C). Calcifications are common and correspond to matrix calcifications or actual endochondral ossifications at the periphery of cartilage lobules (Fig. 3-13). Enchondromas located in the short tubular bones of hands and feet may exhibit increased cellularity, plump nuclei, and occasionally even binucleated cells.
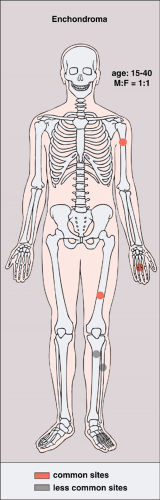 Figure 3-5 Enchondroma: skeletal sites of predilection, peak age range, and male-to-female ratio. |
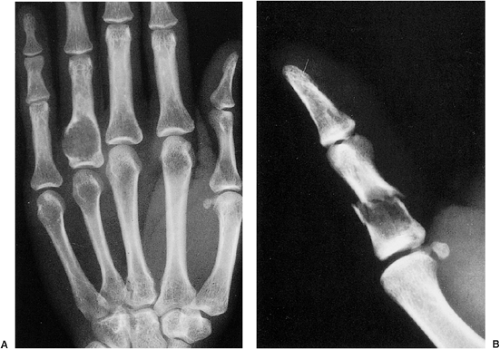 Figure 3-6 Enchondroma in short tubular bones. A: Typical, purely radiolucent lesion at the base of the proximal phalanx of the ring finger in a 37-year-old woman. Note the marked attenuation of the ulnar side of the cortex. B: Radiolucent lesion at the base of the proximal phalanx of the thumb with a pathologic fracture. |
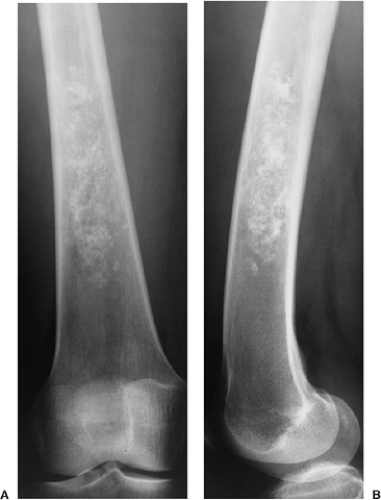 Figure 3-7 Enchondroma in a long bone. Anteroposterior (A) and lateral (B) radiographs of the femur show a radiolucent lesion with “popcorn” calcifications. |
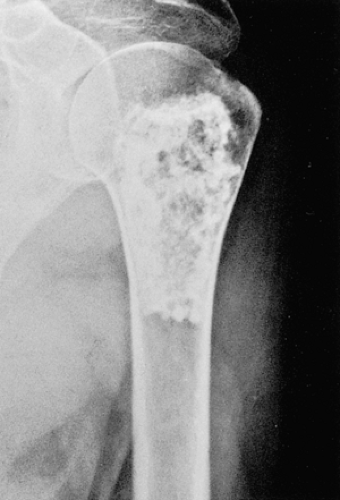 Figure 3-8 “Calcifying enchondroma.” A heavily calcified lesion in the proximal humerus of a 58-year-old woman. |
Only a few cytogenetic studies with similar results have been reported on enchondromas, periosteal chondromas, and soft tissue chondromas, which are therefore discussed together. Flow cytometry studies revealed DNA-diploid peaks, although DNA-aneuploidy has rarely been observed (2,35). Most chondromas contain clonal chromosomal abnormalities involving chromosomes or chromosomal regions 4q, 5, 7, 11,14q, 16q22–q24, 20, and particularly rearrangements of chromosome 6 and 12q12–q15 (8). Tumor-specific anomalies, however, were not detected.
Periosteal (Juxtacortical) Chondroma
Periosteal or juxtacortical chondroma, a slow-growing lesion, affects the surface of a bone in or beneath the periosteum. Preferred sites include the proximal humerus, femur, tibia, and the phalanges of the hand (52). This is an uncommon neoplasm, accounting for about only 0.66% of bone tumors in the Mayo Clinic series (56). Periosteal chondroma was first described by Lichtenstein and Hall in 1952 (39). It occurs in children and adults (most commonly in the second and third decades), who usually present with a history of pain and local tenderness, often accompanied by swelling at the involved site.
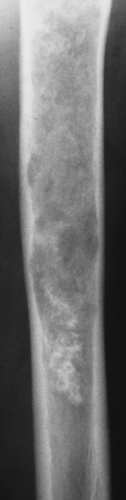 Figure 3-9 Enchondroma. A radiolucent lesion exhibits central calcifications and scalloped inner cortical margins, reflecting the lobular growth pattern of cartilage. |
The radiographic appearance is rather characteristic (6,65). The lesion is small and is located on the surface of the bone (Fig. 3-14). As it enlarges, it erodes the underlying cortex in a saucer-like fashion, displaying a sclerotic rim and producing a buttress of periosteal new bone formation (15). The lesion may contain calcifications (Fig. 3-15). CT may show the scalloped cortex, matrix calcification (Fig. 3-16), and cortical shell (28,36). It also may demonstrate the separation of a lesion from the medullary cavity, an important feature in differentiation from osteochondroma (26). MRI findings correspond to radiographic findings, depicting the cartilaginous soft tissue component (76). If periosteal chondroma affects the medullary canal, MRI may be useful in depicting the extent of involvement (Fig. 3-17). Fat suppression
or enhanced gradient echo sequences may improve tumor–marrow contrast (5). The potential pitfall of MRI is marrow edema mimicking tumor invasion or vice versa (76). Unlike enchondroma and osteochondroma, periosteal chondroma may continue to grow after skeletal maturation (20). Some lesions may attain a large size (up to 6 cm) and may resemble osteochondromas (26) (Fig. 3-18; also see Fig. 3-34). Some lesions may mimic an aneurysmal bone cyst. Very rarely the lesion may encase itself intracortically, thus mimicking other intracortical lesions such as intracortical angioma, intracortical fibrous dysplasia, intracortical bone abscess (1), or osteoid osteoma (63,67).
or enhanced gradient echo sequences may improve tumor–marrow contrast (5). The potential pitfall of MRI is marrow edema mimicking tumor invasion or vice versa (76). Unlike enchondroma and osteochondroma, periosteal chondroma may continue to grow after skeletal maturation (20). Some lesions may attain a large size (up to 6 cm) and may resemble osteochondromas (26) (Fig. 3-18; also see Fig. 3-34). Some lesions may mimic an aneurysmal bone cyst. Very rarely the lesion may encase itself intracortically, thus mimicking other intracortical lesions such as intracortical angioma, intracortical fibrous dysplasia, intracortical bone abscess (1), or osteoid osteoma (63,67).
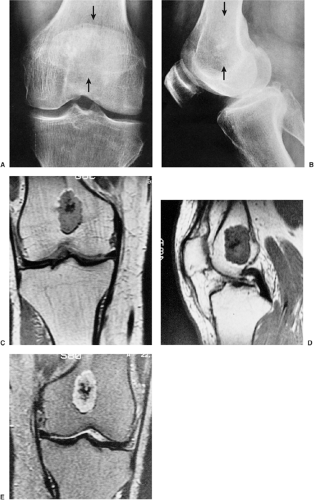 Figure 3-10 Enchondroma: magnetic resonance imaging (MRI). Anteroposterior (A) and lateral (B) radiographs of the knee of a 61-year-old man show a few calcifications in the distal femur (arrows). The nature and extent of the lesion cannot be adequately determined. Coronal (C) and sagittal (D) T1-weighted MRI demonstrates a well-circumscribed, lobulated lesion with intermediate signal intensity. The darker area in the center represents calcifications. E: Coronal T2-weighted image shows the lesion displaying a mixed-intensity signal. The brighter areas represent cartilaginous tumor and the darker areas represent calcifications. |
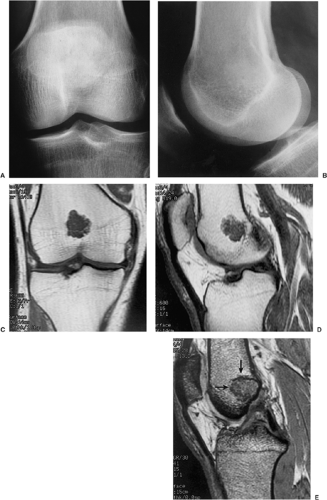 Figure 3-11 Enchondroma: magnetic resonance imaging. Anteroposterior (A) and lateral (B) radiographs show an almost nondiscernible lesion in the distal femur. Coronal (C) and sagittal (D) T1-weighted sequences reveal the full extent of the lesion. E: On T2* (MPGR) image, enchondroma (arrows) is not as bright as on conventional T2-weighted images. |
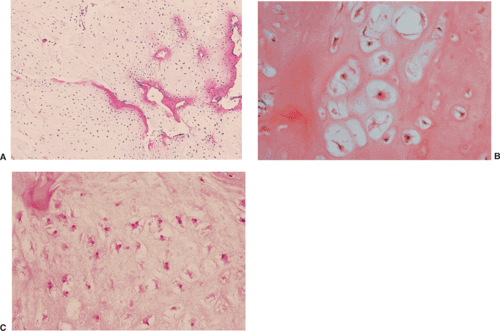 Figure 3-12 Histopathology of enchondroma. A: At low magnification the lesion exhibits hyaline cartilage of low to moderate cellularity (hematoxylin and eosin, original magnification ×50). B: At higher magnification the chondrocytes with darkly stained nuclei are seen to be located in the lacunae (hematoxylin and eosin, original magnification ×100). C: Occasionally binucleated cells may be present (hematoxylin and eosin, original magnification ×150) (C, courtesy of Dr. K. K. Unni, Rochester, Minnesota.) |
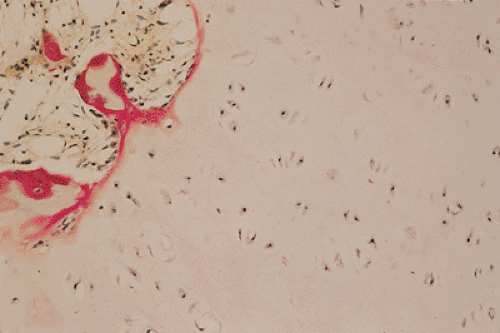 Figure 3-13 Histopathology of enchondroma. Cartilage lobules are surrounded by a narrow rim of bone, representing endochondral ossification (hematoxylin and eosin, original magnification ×250). |
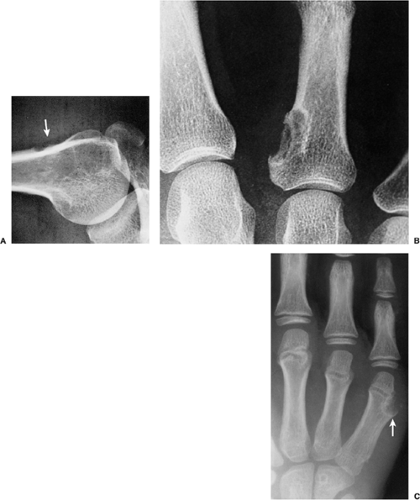 Figure 3-14 Periosteal chondroma. A: Radiolucent lesion erodes the external surface of the cortex of the proximal humerus (arrow) of a 24-year-old man. B: Well-defined, saucer-like erosion of the cortex of the proximal phalanx is characteristic of periosteal chondroma. (Reprinted from Bullough PG. Atlas of orthopedic pathology, 2nd ed. New York: Gower, 1992:16.22.) C: Metaphyseal periosteal chondroma affecting the fifth metacarpal bone erodes the cortex and evokes the buttress of periosteal reaction (arrow). |
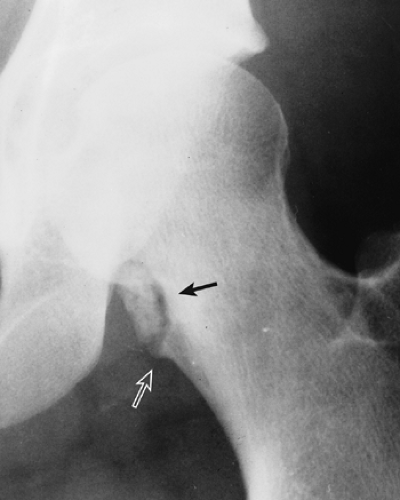 Figure 3-15 Periosteal chondroma. A lesion displaying calcifications erodes the medial cortex of the femoral neck (arrow). The buttress of a periosteal reaction seen at the inferior border of the lesion (open arrow) is characteristic of periosteal chondroma. |
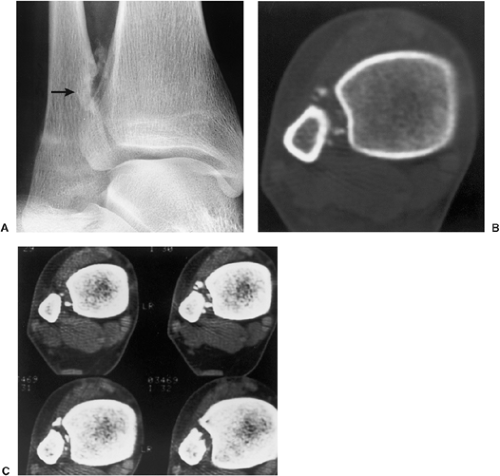 Figure 3-16 Periosteal chondroma: computed tomography (CT). A: Radiograph shows a lesion with calcifications eroding the medial cortex of the distal fibula (arrow). CT using a bone window (B) and a soft tissue window (C) better demonstrates the extent of the lesion and the distribution of the calcifications. |
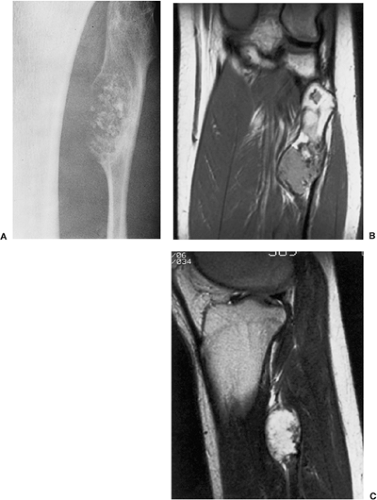 Figure 3-17 Periosteal chondroma: magnetic resonance imaging (MRI). A: Large lesion blends imperceptibly with the medial fibular cortex and extends into the medullary cavity. In such a presentation, differential diagnosis must include enchondroma protuberans and chondrosarcoma. Coronal proton-density (SE, TR 2000, TE 19) (B) and sagittal T2-weighted (SE, TR 2000, TE 70) (C) MRI shows the lesion’s extension into the bone marrow. |
On histologic examination, the findings are identical to those of enchondroma, although the lesion sometimes exhibits a higher level of cellularity, occasionally with atypical and binucleated cells (Fig. 3-19). Well-formed lamellar bone or fibrous connective tissue separates the cartilaginous nodules, and in some instances deposition of calcium can be observed (16).
Enchondromatosis, Ollier Disease, and Maffucci Syndrome
Enchondromatosis is a condition marked by multiple enchondromas, usually involving the metaphyses and diaphyses (21). If skeletal involvement is extensive (42), and particularly if the involvement is unilateral [resembling the original case described by Ollier (50,58,59,72)], the term “Ollier disease” is applied
(68). There is no hereditary or familial tendency in this disorder, which some investigators consider to be developmental rather than neoplastic, classifying it as a form of bone dysplasia caused by failure of normal endochondral ossification (45). The femur, tibia, and ilium are the most commonly affected bones (72), followed by the phalanges, metacarpals, and metatarsals. Much less often the lesions involve the facial bones, skull, spine, carpal, and tarsal bones (17,48) (Fig. 3-20).
(68). There is no hereditary or familial tendency in this disorder, which some investigators consider to be developmental rather than neoplastic, classifying it as a form of bone dysplasia caused by failure of normal endochondral ossification (45). The femur, tibia, and ilium are the most commonly affected bones (72), followed by the phalanges, metacarpals, and metatarsals. Much less often the lesions involve the facial bones, skull, spine, carpal, and tarsal bones (17,48) (Fig. 3-20).
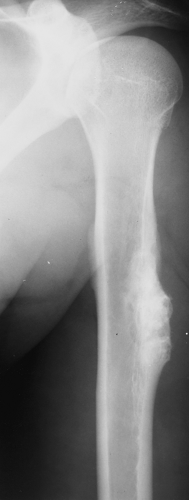 Figure 3-18 Periosteal chondroma. Large lesion eroding the cortex of the proximal humerus resembles a sessile osteochondroma. Note the periosteal reaction and separation of the tumor from the medullary cavity by a cortex, features that help in the differentiation from osteochondroma. (Courtesy Dr. K. K. Unni, Rochester, Minnesota.) |
Maffucci syndrome is a congenital, nonhereditary disorder, characterized by enchondromatosis and soft tissue angiomatosis (hemangiomatosis) (19). The latter may occur anywhere in the skin and subcutaneous tissue. The hemangiomas are usually cavernous and may be unilateral or bilateral, localized or extensive. The skeletal lesions in Maffucci syndrome have predilection for tubular bones and have the same distribution as those in Ollier disease, with a strong predisposition for one side of the body (75). The most frequent sites of enchondromas are the metacarpals and the phalanges of the hands (37).
Recent investigations suggest that Ollier disease and Maffucci syndrome represent two entities within a continuum of the disease multiple enchondromatosis and that both conditions bear the risk of mesodermal as well as nonmesodermal malignancy (27).
Clinical Presentation
The condition has a strong preference for involvement of one side of the body. The clinical manifestations, such as knobby swellings of the digits or gross disparities in limb length, are commonly recognized during childhood and adolescence. The affected child often begins to limp during the second year of life as a disparity in leg length becomes apparent (long bones that are affected are always shorter because the growth plate is involved). Pain is uncommon and when present is usually due to a pathologic fracture (52). Ollier disease is often arrested at puberty but may occasionally progress after that time. The most common and severe complication of Ollier disease is malignant transformation of one or more enchondromas to chondrosarcoma (34,41) (see Figs. 3-118 and 3-119). Schajowicz (68) believed that about 30% of the patients with Ollier disease will develop chondrosarcoma, but Jaffe (30) and Mirra et al. (49) have estimated that risk to be as high as 50%. However, because patients with enchondromatosis or Ollier disease may develop hundreds of enchondromas, the statistical risk of one given lesion to undergo malignant degeneration is probably not higher than in solitary enchondroma. Unlike solitary enchondromas, in patients with enchondromatosis even lesions in the short tubular bones may undergo sarcomatous changes. This is particularly true in patients with Maffucci syndrome (73).
Imaging
The radiographic appearance of enchondromatosis involving the hands and feet is quite characteristic. Radiolucent masses of cartilage with foci of calcifications closely resembling solitary enchondromas markedly deform the bones (Fig. 3-21). Enchondromas in this location may be intracortical and periosteal (50). They sometimes protrude from the shaft of the short or long bone, thus resembling osteochondroma (Fig. 3-22). However, unlike osteochondroma, these protruding enchondromas have neither a cartilaginous cap nor a bony stalk. Also unlike osteochondroma, the protrusions do not point away from the adjacent joint (50). In the long bones, columns of radiolucent streaks extend from the growth plate into the diaphysis. Coalescence of metaphyseal enchondromas often causes asymmetric enlargement of the long bones and flaring of the metaphyses. Interference with the growth plate causes foreshortening and deformity of the bones (Fig. 3-23). In the pelvis, and particularly in the iliac bones, a fan-like linear pattern is characteristic (Fig. 3-24). Maffucci syndrome is recognized radiographically by multiple calcified phleboliths in addition to the typical alterations of enchondromatosis (Fig. 3-25) (45).
Histopathology
Histologically, the lesions in enchondromatosis are essentially indistinguishable from those of solitary enchondromas, although they sometimes tend to be predominantly myxoid, more cellular, and may exhibit more cellular atypia (Fig. 3-26). Mitoses and prominent nucleoli are uncommon, and anaplasia is not observed (13). However, binucleated cells are often present.
Both Ollier disease and Maffucci syndrome occur sporadically although familial cases have been described (44). In a molecular genetic study Hopyan et al. (29) demonstrated a mutation in the gene of the parathyroid
hormone receptor 1 (PTHR1), one germline and one somatic mutation, in two of six patients with Ollier disease leading to an abnormal signaling and, in transgenic mice, to enchondroma-like lesions. However, in an extensive study on 23 enchondromas and 18 chondrosarcomas from 31 patients with enchondromatosis, Rozeman et al. (66) could not confirm these findings or demonstrate any different mutation in the PTHR1 gene.
hormone receptor 1 (PTHR1), one germline and one somatic mutation, in two of six patients with Ollier disease leading to an abnormal signaling and, in transgenic mice, to enchondroma-like lesions. However, in an extensive study on 23 enchondromas and 18 chondrosarcomas from 31 patients with enchondromatosis, Rozeman et al. (66) could not confirm these findings or demonstrate any different mutation in the PTHR1 gene.
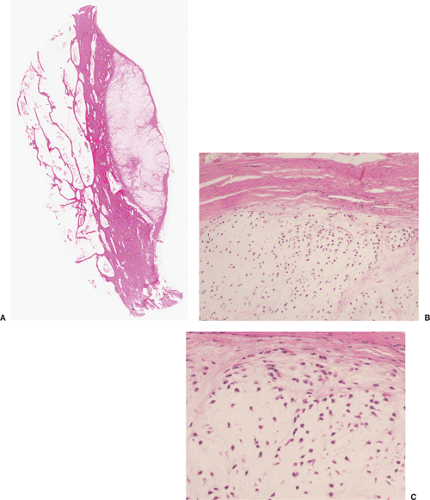 Figure 3-19 Histopathology of periosteal chondroma. A: At the interface with adjacent cortex, the cartilaginous tumor (right) erodes the bone (hematoxylin and eosin, whole-mount section). B: The tumor tissue is more cellular than conventional enchondroma, and some cells appear atypical (hematoxylin and eosin, original magnification ×100). C: Hyaline cartilage with a rather dense cell population, slight cell pleomorphism, and signs of cell proliferation. These histologic variations are compatible with a benign cartilage lesion in a periosteal location (hematoxylin and eosin, original magnification ×200). |
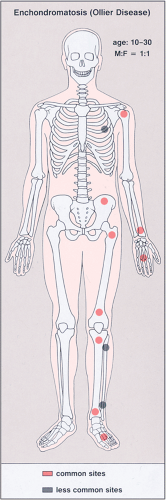 Figure 3-20 Enchondromatosis (Ollier disease): skeletal sites of predilection, peak age range, and male-to-female ratio. |
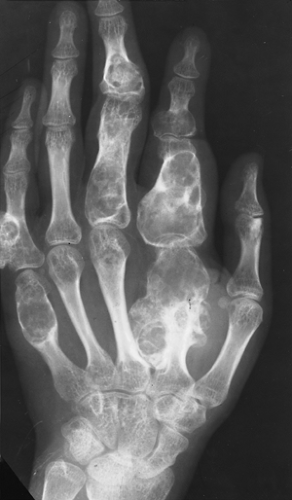 Figure 3-21 Enchondromatosis (Ollier disease). Large, lobulated cartilaginous masses markedly deform the bones of the hand. |
Only two other genetic analyses in chondrosarcomas developing in enchondromatosis have been reported so far. Using cytogenetics, Ozisik et al. (60) showed an interstitial deletion [del(1)(p11p31.2)] at the short arm of chromosome 1, whereas Bovée et al. (7) observed loss of heterozygosity (LOH: loss of one allele in the tumor DNA compared to normal germline DNA of an individual) at 13q14, the locus of the tumor suppressor gene RB1 (retinoblastoma gene), and at 9p21 among others where two cell cycle regulating genes (p15/p16) are located.
Differential Diagnosis
Radiology
The main differential diagnosis in solitary enchondroma, particularly when it is located in long bones
and exhibits substantial calcifications (“calcifying enchondroma”), is medullary bone infarct (Fig. 3-27). At times the two lesions are difficult to distinguish from one another because of similar calcifications, particularly when the enchondroma is small. A number of radiographic features can be helpful in the differential diagnosis. These include lobulation of the margins of an enchondroma, punctate or annular calcifications within the matrix, and absence of the peripheral sclerotic fibro-osseous wall commonly observed in bone infarcts (18). Mineral deposits also tend to exhibit a more central distribution, and the calcifications often have a
markedly stippled appearance (Fig. 3-28). Although the ring-like calcifications typical of enchondroma are occasionally present in medullary bone infarct, they are usually larger and have thicker walls. As a rule, bone infarct, unlike enchondroma, does not expand bone contours (51) except in rare cases of encystification (57) (see Fig. 7-54). The distribution of the medullary bone infarcts that commonly affect the proximal and distal femur and the proximal tibia may be helpful in the differential diagnosis.
and exhibits substantial calcifications (“calcifying enchondroma”), is medullary bone infarct (Fig. 3-27). At times the two lesions are difficult to distinguish from one another because of similar calcifications, particularly when the enchondroma is small. A number of radiographic features can be helpful in the differential diagnosis. These include lobulation of the margins of an enchondroma, punctate or annular calcifications within the matrix, and absence of the peripheral sclerotic fibro-osseous wall commonly observed in bone infarcts (18). Mineral deposits also tend to exhibit a more central distribution, and the calcifications often have a
markedly stippled appearance (Fig. 3-28). Although the ring-like calcifications typical of enchondroma are occasionally present in medullary bone infarct, they are usually larger and have thicker walls. As a rule, bone infarct, unlike enchondroma, does not expand bone contours (51) except in rare cases of encystification (57) (see Fig. 7-54). The distribution of the medullary bone infarcts that commonly affect the proximal and distal femur and the proximal tibia may be helpful in the differential diagnosis.
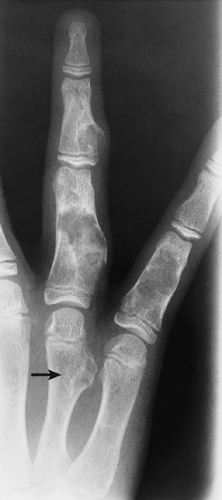 Figure 3-22 Enchondromatosis (Ollier disease). Multiple lesions are observed in the phalanges and metacarpals. The intracortical lesion in the metaphysis of the fourth metacarpal protrudes from the bone (arrow), thus resembling an osteochondroma. |
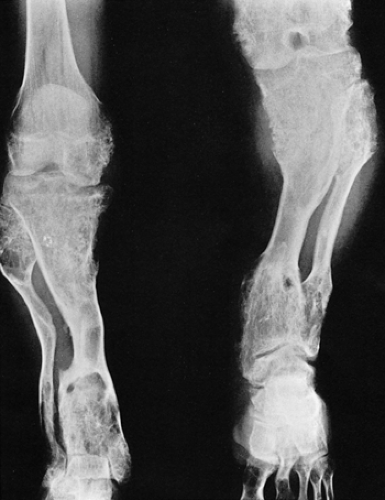 Figure 3-23 Enchondromatosis (Ollier disease). Anteroposterior radiograph of both legs of a 17-year-old boy shows growth stunting and marked deformities of the tibiae and fibulae. |
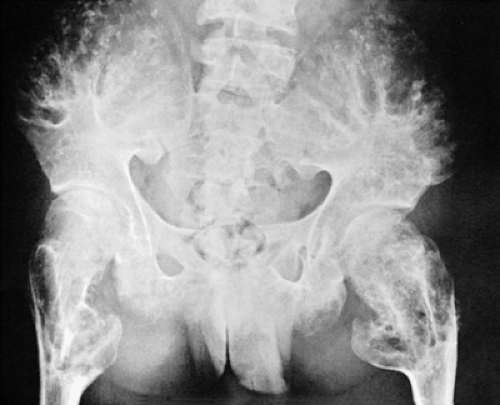 Figure 3-24 Enchondromatosis (Ollier disease). Anteroposterior radiograph of the pelvis demonstrates crescent-shaped and ring-like calcifications in tongues of cartilage extending from the iliac crests and the proximal femora, classic features of Ollier disease. |
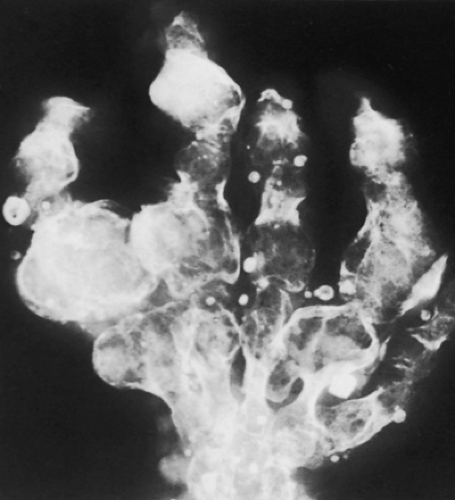 Figure 3-25 Maffucci syndrome. Radiograph of the hand reveals typical changes of enchondromatosis, accompanied by calcified phleboliths in soft tissue hemangiomas. (Reprinted from Bullough PG. Atlas of orthopedic pathology, 2nd ed. New York: Gower, 1992:14.9.) |
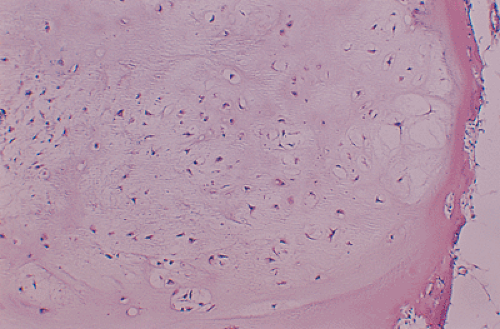 Figure 3-26 Histopathology of enchondromatosis. Lobular and cellular appearance of cartilaginous nodules. These lesions usually exhibit more cellularity than is seen in solitary enchondroma (hematoxylin and eosin, original magnification ×100). |
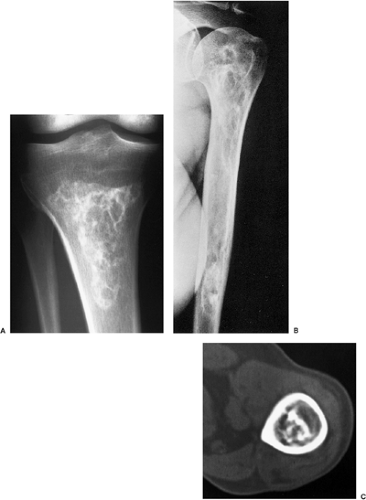 Figure 3-27 Medullary bone infarct. A: Anteroposterior radiograph of the proximal tibia shows the typical coarse calcifications of bone infarct. Note the sharply defined peripheral margin separating infarcted from viable bone and the lack of annular and comma-shaped calcifications characteristic for cartilage tumor (compare with Fig. 3-28). B: In a medullary bone infarct, seen here in the proximal humerus of a 36-year-old man with sickle cell disease, there is no endosteal scalloping of the cortex, and the calcified area is surrounded by a thin, dense sclerotic rim, the hallmark of a bone infarct. C: In another patient with a bone infarct in the distal femur, a computed tomography section reveals central calcifications and no endosteal scalloping of the cortex. |
The most difficult task for the radiologist is to distinguish a large solitary enchondroma from a slowly growing low-grade chondrosarcoma (53). One of the most significant findings pointing to a chondrosarcoma in the early stage of development is localized thickening of the cortex (Fig. 3-29). Deep endosteal scalloping (greater than two thirds of cortical thickness) is another sign of possible malignancy (53). The size of the lesion should also be taken into consideration: lesions longer than 4 cm are suggestive of malignancy (Fig. 3-30). In advanced lesions, destruction of the cortex, periosteal reaction of aggressive type, and the presence of a soft tissue mass are the hallmarks of malignancy.
It is equally important to distinguish sarcomatous transformation in patients with Ollier disease and Maffucci syndrome. Radiography will reveal intralesional radiolucencies and cortical destruction associated with a soft tissue mass (see Fig. 3-118).
If enchondroma, particularly in a short, tubular bone, extends to the articular end of the bone, giant cell tumor should be included in the differential diagnosis. The latter contains no calcifications, an important differential clue. However, some enchondromas may not display visible calcifications. In addition, giant cell tumor usually lacks a sclerotic border. In the long, tubular bones, this distinction is much easier because enchondroma, unlike giant cell tumor, only rarely extends into the articular end of bone (Fig. 3-31). Enchondroma may mimic solitary bone cyst, although the latter is a rare occurrence in the short, tubular bones. If an enchondroma is found in the distal phalanx of the hand and does not contain calcifications, an epidermoid cyst is a differential possibility.
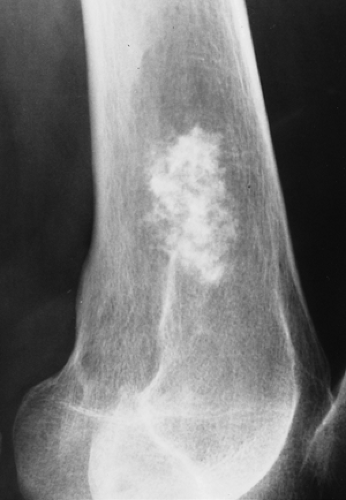 Figure 3-28 Enchondroma. Typical appearance of annular and stippled calcifications within a radiolucent lesion. |
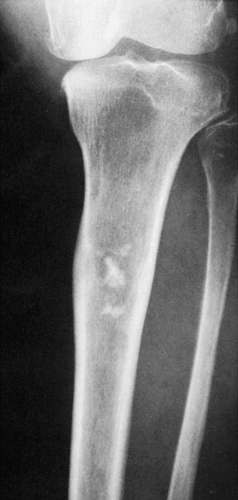 Figure 3-29 Low-grade chondrosarcoma. A 48-year-old woman presented with pain in the upper leg. Radiograph shows a radiolucent lesion in the proximal tibia with a wide zone of transition and central calcifications. Note the thickening of the cortex, a feature that distinguishes chondrosarcoma from similarly appearing enchondroma. |
An intracortical enchondroma, or an enchondroma that causes nonsymmetric cortical expansion, may be mistaken for osteochondroma (see Fig. 3-22). However, the
distribution of the mineral matrix visualized within enchondroma usually helps to distinguish the two entities.
distribution of the mineral matrix visualized within enchondroma usually helps to distinguish the two entities.
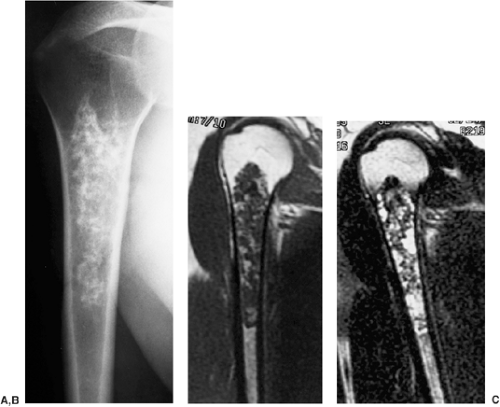 Figure 3-30 Low-grade chondrosarcoma. A: Conventional radiograph in a 48-year-old man shows a cartilaginous lesion in the proximal right humerus. Although the cortex is not thickened, the length of the lesion (15 cm) suggests malignancy. Coronal T1-weighted (B) and T2-weighted (C) magnetic resonance imaging shows heterogeneous signal intensity, which may be seen in both enchondroma and chondrosarcoma. On biopsy, the tumor proved to be a low-grade chondrosarcoma. |
Enchondroma protuberans may create a problem in diagnosis (10). Enchondroma protuberans has been defined as an exophytic enchondroma of a long bone, although it may affect a rib as well (31). It arises in the medullary cavity but penetrates the cortex, forming a prominent exophytic mass on the surface of bone, and thus can mimic any surface lesion (e.g., osteoma, osteochondroma, periosteal chondroma, myositis ossificans). It also should be distinguished from chondrosarcoma, either primary or one developing in a preexisting enchondroma. Further distinction must be made from the simultaneous occurrence of an osteochondroma and an enchondroma in the same bone (64) (Fig. 3-32).
Ollier disease almost never creates a diagnostic problem. Only occasionally the Jansen type of metaphyseal chondrodysplasia, a skeletal disorder manifested by radiologically detectable, generalized, symmetric metaphyseal abnormalities, may mimic the former condition. Widening and flaring of the metaphyses and irregularity of the zones of provisional calcification may be mistaken for enchondromas. However, other features of this dysplasia, such as rhizomelic
short-limb dwarfism, enlarged joints, craniofacial changes including large skull with frontonasal hyperplasia, and hyperostosis of the cranial vault and base of the skull, should be helpful in arriving at the correct diagnosis.
short-limb dwarfism, enlarged joints, craniofacial changes including large skull with frontonasal hyperplasia, and hyperostosis of the cranial vault and base of the skull, should be helpful in arriving at the correct diagnosis.
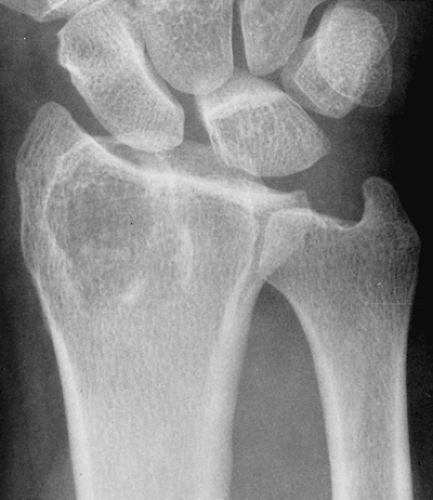 Figure 3-31 Enchondroma resembling giant cell tumor. The lesion extends into the articular end of the distal radius and therefore may mimic a giant cell tumor. However, unlike the latter, there are a few faintly visualized calcifications and a sclerotic margin. |
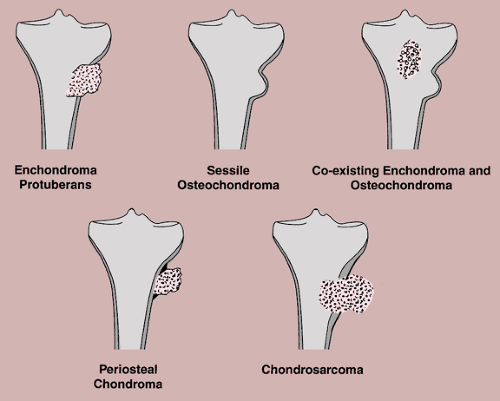 Figure 3-32 Differential diagnosis of enchondroma protuberans. |
Periosteal chondroma should be radiographically differentiated from a variety of other periosteal lesions (e.g., periosteal ganglion, periosteal Ewing sarcoma), as well as from myositis ossificans and tumoral calcinosis. Features of myositis ossificans have been described in the section on extraskeletal osteosarcoma. The pattern of zoning phenomenon is diagnostic of the former lesion. Features of tumoral calcinosis (Fig. 3-33) have been described in the section on synovial sarcoma (see Chapter 9).
A large periosteal chondroma may closely resemble sessile osteochondroma and vice versa. The key to distinguishing between these two lesions is the separation of periosteal chondroma from the medullary portion of the bone by intervening cortex (26) (Fig. 3-34). Conversely, in a typical osteochondroma the cortex of the host bone merges with the cortex of the lesion, and continuity exists between the medullary cavity of the host bone and the exostosis (see Fig. 3-38B). Another useful characteristic is the value of attenuation coefficient of the lesion, as determined from CT scans: the base of the osteochondroma, composed
mainly of trabecular bone, shows higher numbers for this variable than does the cartilaginous matrix of periosteal chondroma.
mainly of trabecular bone, shows higher numbers for this variable than does the cartilaginous matrix of periosteal chondroma.
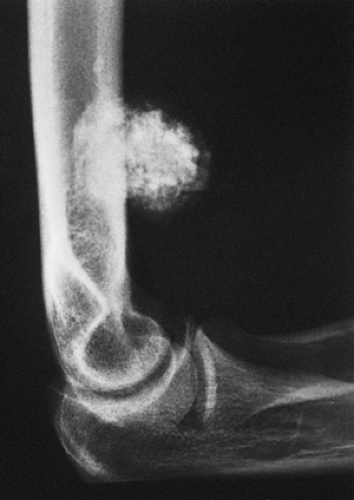 Figure 3-33 Tumoral calcinosis. Although soft tissue calcification may look very similar to that of periosteal chondroma, there is no bone erosion and no periosteal buttress formation. |
Pathology
The main problem is to distinguish benign enchondroma from low-grade chondrosarcoma. In the majority of cases, particularly if the histologic picture is typical and the lesion is not cellular, this distinction does not create a problem (see Figs. 3-2 and 3-3). However, if the lesion is cellular, and particularly if it affects the long bones, (Fig. 3-35), the pathologist must take care not to confuse an enchondroma with a low-grade chondrosarcoma and vice versa.
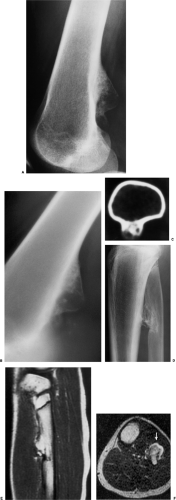 Figure 3-34 Periosteal chondroma resembling osteochondroma. A: Lateral radiograph shows a bony excrescence arising from the posterior cortex of the distal femur that looks like a sessile osteochondroma B: Conventional tomography shows calcifications at the base of the lesion and lack of interruption of the posterior cortex of the femur. C: Computed tomography section demonstrates lack of communication between the medullary portion of the femur and the lesion, thus excluding the diagnosis of osteochondroma. (A and C, reprinted with permission from Greenspan et al. Periosteal chondroma masquerading as osteochondroma. Can Assoc Radiol J 1993;44:205–210.) D: In another patient the lateral radiograph of the proximal leg demonstrates a lesion in the fibula that may represent either a sessile osteochondroma or a periosteal chondroma. E: Sagittal T1-weighted (SE, TR 600, TE 20) magnetic resonance imaging (MRI) reveals that the lesion is brighter than the fibula’s bone marrow and the anterior fibular cortex does not interrupt medullary continuity, suggesting a diagnosis of osteochondroma. F: This diagnosis is further confirmed with axial T2-weighted (SE, TR 2000, TE 80) MRI that shows unequivocally not only the merging of the medullary portions of the lesion and the host bone, but a cartilaginous cap (bright signal), characteristic of osteochondroma (arrow). |
Cell atypias and uneven distribution of cells may be found to a higher degree in enchondromas of the hands and feet without prognostic significance. However, when an enchondroma is located in long, tubular bones, particularly in the humerus and femur, or in bones of the trunk, slight histologic atypias have prognostic significance. From observations of large series over long periods it is known that tumors with these characteristics may recur once or several times and may progress to a chondrosarcoma of intermediate or high
grade, or even to a dedifferentiated chondrosarcoma. Furthermore, enchondroma lobules are often encased by newly formed lamellar bone.
grade, or even to a dedifferentiated chondrosarcoma. Furthermore, enchondroma lobules are often encased by newly formed lamellar bone.
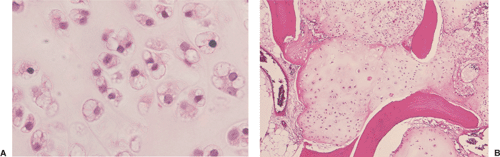 Figure 3-35 Low-grade chondrosarcoma. A: Chondrocytes with a subtle degree of pleomorphism and abnormal nuclei with open chromatin and visible nucleoli are irregularly distributed within a chondroid matrix (hematoxylin and eosin, original magnification ×100). B: Destruction of the bone trabeculae surrounded by chondroblastic tumor tissue that infiltrates the marrow spaces, and a dense arrangement of chondrocytes correspond to grade 2 tumor (hematoxylin and eosin, original magnification ×50). |
The microscopic distinction between benign cartilage of enchondromatosis and low-grade chondrosarcoma is difficult (16). Necrosis and myxoid stroma strongly favor the diagnosis of chondrosarcoma (23).
Histologically, periosteal chondroma must be differentiated from periosteal chondrosarcoma (see Figs. 3-112 and 3-113) and periosteal osteosarcoma (see Figs. 2-109 and 2-110). The latter lesion, however, is usually grade 2 or 3, exhibits a diaphyseal location, and contains osteoid or, less commonly, mature bone.
The radiologic and pathologic differential diagnosis of enchondroma and periosteal chondroma is depicted in Figure 3-36.
Soft Tissue Chondroma
These rare lesions occur preferentially in the soft tissues of hands and feet. A soft tissue chondroma located close to a joint is known as a paraarticular chondroma (22,47). Because these lesions exhibit a wide range of cytologic variations, as in the case of cartilage tumors of bone, they are commonly misdiagnosed as chondrosarcomas (18). They have no predilection for either gender and most patients are 20 years or older, with a mean occurrence during the fourth and fifth decades (9). These lesions are usually small, ranging from 2 to 4 cm. On radiographic studies they present as well-defined soft tissue masses, often (33% to 77%) with calcifications similar to those seen in enchondroma (78). They are not attached to the periosteum or cortex of a bone and are not found within the confines of a joint capsule or tendon sheath (3,12,77). On histopathologic examination soft tissue chondromas consist of masses of hyaline cartilage, which are commonly lobulated and sometimes partially myxoid (18). In addition, some lesions are hypercellular and exhibit hyperchromatic nuclei. These features, which are also seen in enchondromas of small tubular bones, are not indicative of malignancy. Soft tissue chondromas may contain focal areas of fibrosis, hemorrhage, necrosis, calcification, ossification, or granuloma formation (3,77). Electron microscopy shows typical features of cartilage cells, with abundant rough endoplasmic reticulum, free ribosomes (55), and short irregular microvillous processes surrounded by aggregates of calcium crystals (9).
Recently, nonrandom clonal alterations of chromosomes 6, 11, and 12 have been reported in soft tissue chondromas, including supernumerary ring chromosomes containing material from chromosome 12 (14,43,71). Molecular analysis has shown that the so-called HMGA2 gene located at 12q15 (high mobility group gene, coding for a small non-histone-chromatin-associated protein implicated in the architectural organization of chromatin, thus influencing transcription and playing a role in growth and development) appears to be involved in soft tissue chondroma and other cartilaginous tumors, and also in lipomas (11,40).
Differential Diagnosis
Radiology
The main differential possibility is periosteal chondroma. This lesion is, however, attached to the periosteum or cortex and usually produces a cortical erosion and buttress of periosteal reaction. Myositis ossificans can result in a mineralized mass; however, it exhibits a classic zoning phenomenon, whereas calcifications in the soft tissue chondroma are distributed in a haphazard pattern. Synovial chondromatosis is a condition affecting joints and tendon sheaths, and CT or MRI is usually able to identify intra-articular or within the tendon sheath location. Benign mesenchymoma of soft tissue may contain calcifications. These lesions are composed primarily of mature hyaline cartilage. The difference between soft tissue chondroma and mesenchymoma is that the latter may also contain adipose and vascular elements (46). Lesions reported in the literature as soft tissue osteochondromas may appear similar to soft tissue chondromas, although in addition to calcifications they also may contain bony elements (38).
Tumoral calcinosis usually presents on radiography as a well-defined lobulated calcific mass, exhibiting a layering effect when imaged with a horizontal beam (4,61) (see Fig. 3-33).
Soft tissue chondrosarcomas are rare lesions (77). Unlike soft tissue chondromas they almost never occur in the hands and feet, their preferred sites being in the proximal parts of extremities and buttocks. Synovial sarcoma exhibits a soft tissue mass and, in approximately 25% to 30% of reported cases, calcifications. It is usually accompanied by destruction of adjacent bones.
Pathology
In general, soft tissue chondromas are so characteristic that problems in differential diagnosis rarely arise, although hypercellular lesions with hyperchromatic nuclei may be confused with chondrosarcomas. However, extraskeletal chondrosarcomas are almost always of myxoid or mesenchymal type, are rarely located in the hands and feet, and contain a prevailing small-cell component. Because no extraosseous low-grade chondrosarcoma composed of pure hyaline cartilage has yet been reported, this diagnosis can be excluded (32). Nevertheless, soft tissue extensions of intraosseous grade 1 chondrosarcoma must be ruled out.
Calcifying aponeurotic fibroma, mainly occurring in the hands and feet of children, contains foci of cartilage. However, in contrast to well-demarcated and lobulated soft tissue chondromas, these lesions are ill-defined and tend to infiltrate the surrounding soft tissues. Synovial chondromatosis is, by definition, located within synovial tissue and consists of multiple cartilaginous nodules. Myxoid changes in soft tissue chondroma may lead to the erroneous diagnosis of extraskeletal myxoid chondrosarcoma, which, however, preferentially occurs in the lower extremities and not in the hands and feet. In addition, this tumor is larger and more cellular, containing round cells with scant eosinophilic cytoplasm
arranged in cords and clusters. These features are absent in soft tissue chondroma (77).
arranged in cords and clusters. These features are absent in soft tissue chondroma (77).
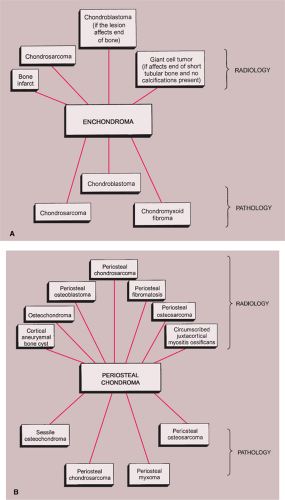 Figure 3-36 Radiologic and pathologic differential diagnosis of enchondroma (A) and periosteal chondroma (B). |
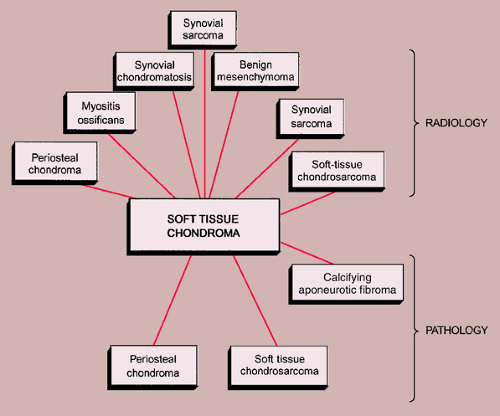 Figure 3-37 Radiologic and pathologic differential diagnosis of soft tissue chondroma. |
The radiologic and pathologic differential diagnosis of soft tissue chondroma is shown in Figure 3-37.
Osteochondroma (Osteocartilaginous Exostosis)
Clinical Presentation
Osteochondroma, the most common benign bone lesion (representing about 45% of all benign bone tumors and 12% of all bone tumors) (13), is a cartilage-capped bony projection on the external surface of a bone. Usually diagnosed before the third decade, it most commonly involves the metaphyses of long bones, particularly around the knee and the proximal humerus. In general, the lower extremities are more often affected than the upper extremities. The flat bones, including the scapula, ilium, and clavicle, are much less commonly affected (Fig. 3-38). The lesion, which possesses its own “growth plate,” usually stops growing at skeletal maturity. Only sporadic cases of spontaneous resolution have been reported (113). Osteochondroma is asymptomatic unless it causes pressure on adjacent muscles, nerves, or blood vessels (97), as well as on adjacent bone, occasionally with consequent fracture (116). Other complications have been reported, such as fracture of the lesion itself (84) and inflammatory changes of the bursa exostotica covering the cartilaginous cap (79,86,92) (see Figs. 3-54 and 3-55). Malignant transformation to chondrosarcoma is very rare, occurring in less than 1% of solitary lesions (see Fig. 3-52). Pain (in the absence of a fracture, bursitis, or pressure on nerves) and a growth spurt or continued growth of the lesion beyond skeletal maturity are highly suspicious for this complication (110) (Table 3-1).
Imaging
Radiographic presentation of osteochondroma depends on the type of lesion (107). The pedunculated osteochondroma manifests with a slender pedicle, which is usually directed away from the neighboring growth plate or joint (Fig. 3-39A). The sessile osteochondroma exhibits a broad base attached to the cortex (Fig. 3-39B). In either type, the most important identifying features are that the cortex of the host bone merges without interruption with the cortex of the osteochondroma and that the cancellous portion of the lesion is continuous with the medullary cavity of the adjacent diaphysis. Calcifications in the chondroosseous portion of the stalk of the lesion are also typical features (Fig. 3-40). CT
scanning can establish unequivocally the continuity of cancellous portions of the lesion and the host bone (Fig. 3-41). These characteristics distinguish this lesion from the occasionally similar-appearing bone masses of osteoma, juxtacortical osteosarcoma, soft tissue osteosarcoma, and juxtacortical myositis ossificans (see Fig. 2-6). CT or MRI examination demonstrates the cartilaginous cap (51) (Fig. 3-42). On MRI the cartilaginous cap shows a high signal intensity on T2-weighted and gradient echo sequences. A narrow band of low signal intensity surrounding the cap represents the perichondrium (91,102) (Fig. 3-43). Small areas of various degrees of signal void and low signal intensity represent cartilage calcifications. Gadolinium-enhanced MRI may be helpful in demonstrating peripheral enhancement in osteochondroma, corresponding to fibrovascular tissue covering the nonenhancing cartilage cap (91) (Fig. 3-44).
scanning can establish unequivocally the continuity of cancellous portions of the lesion and the host bone (Fig. 3-41). These characteristics distinguish this lesion from the occasionally similar-appearing bone masses of osteoma, juxtacortical osteosarcoma, soft tissue osteosarcoma, and juxtacortical myositis ossificans (see Fig. 2-6). CT or MRI examination demonstrates the cartilaginous cap (51) (Fig. 3-42). On MRI the cartilaginous cap shows a high signal intensity on T2-weighted and gradient echo sequences. A narrow band of low signal intensity surrounding the cap represents the perichondrium (91,102) (Fig. 3-43). Small areas of various degrees of signal void and low signal intensity represent cartilage calcifications. Gadolinium-enhanced MRI may be helpful in demonstrating peripheral enhancement in osteochondroma, corresponding to fibrovascular tissue covering the nonenhancing cartilage cap (91) (Fig. 3-44).
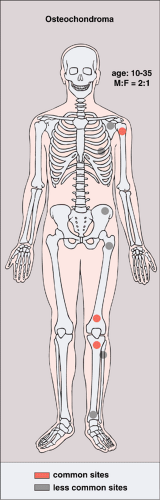 Figure 3-38 Osteochondroma: skeletal sites of predilection, peak age range, and male-to-female ratio. |
Ultrasound (sonography) may also be a valuable modality for measuring the thickness of the cartilaginous cap of osteochondroma (107). In fact, the investigations conducted by Malghem et al. (105) revealed that this technique was more accurate than CT and quite similar to MRI in evaluation of cap thickness. Disadvantages of ultrasound included operator dependence, inability to evaluate deep lesions, and inability to evaluate the osseous component of osteochondroma.
Scintigraphy shows variably increased uptake by the lesion (87). The intensity of activity is directly correlated with the degree of chondral ossification (101). However, the main use of scintigraphy is to search for multiple lesions, when only a single osteochondroma has been discovered by radiography.
Histopathology
Histologically, the cap of the osteochondroma is composed of hyaline cartilage arranged similarly to a growth plate. A zone of provisional calcification is present, corresponding to the areas of calcifications in the chondroosseous portion of the stalk (Fig. 3-45). Deeper parts of the stalk show fatty marrow that merges with normal hematopoietic marrow of the host bone. The thickness of the cartilaginous cap ranges from 1 to 3 mm and rarely up to 1 cm. Greater thickness may imply the possibility of transformation to chondrosarcoma (18). A delicate, fibrous membrane known as the perichondrium, representing a continuation of the periosteum of the adjacent cortex, overlies the cartilaginous cap (see Fig. 3-45D). With age the cap atrophies and in some instances disappears completely (51).
Multiple Hereditary Osteochondromata (Hereditary Osteochondromatosis)
Classified by some authorities among the bone dysplasias, multiple osteochondromata, also known as multiple osteocartilaginous exostoses, familial or hereditary osteochondromatosis, or diaphyseal aclasis, represent an autosomal dominant, hereditary disorder (96), with incomplete penetrance in females (103). Approximately 66% of affected individuals have a positive
family history (81). The specific genetic abnormalities have recently been detected, with three distinct loci on chromosomes 8, 11, and 19 (81,104,117). There is a decided 2:1 male predilection (Fig. 3-46). The lesions are usually discovered at about 2 years of age. The knees, hips, ankles, and shoulders are the most commonly affected sites and growth disturbances are often present, primarily in the forearms and legs (111). There is evidence of defective metaphyseal remodeling, with deformation of affected bones and asymmetric retardation of longitudinal bone growth (16). The radiologic features are similar to those of a solitary osteochondroma; the sessile form of the lesion is more common (Figs. 3-47, 3-48, 3-49, 3-50 and 3-51).
family history (81). The specific genetic abnormalities have recently been detected, with three distinct loci on chromosomes 8, 11, and 19 (81,104,117). There is a decided 2:1 male predilection (Fig. 3-46). The lesions are usually discovered at about 2 years of age. The knees, hips, ankles, and shoulders are the most commonly affected sites and growth disturbances are often present, primarily in the forearms and legs (111). There is evidence of defective metaphyseal remodeling, with deformation of affected bones and asymmetric retardation of longitudinal bone growth (16). The radiologic features are similar to those of a solitary osteochondroma; the sessile form of the lesion is more common (Figs. 3-47, 3-48, 3-49, 3-50 and 3-51).
Table 3-1 Clinical and Radiologic Features Suggesting Malignant Transformation of Osteochondroma | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
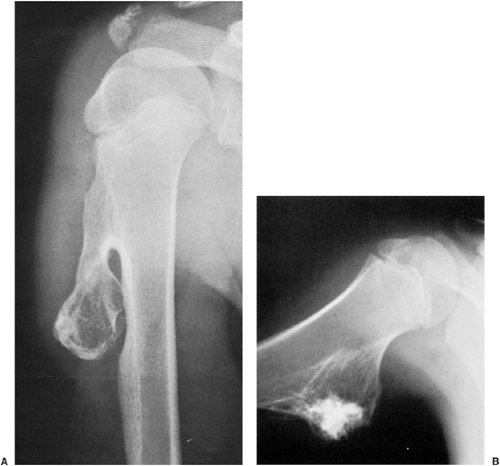 Figure 3-39 Osteochondroma. A: Pedunculated variant. The typical pedunculated type of osteochondroma is seen arising near the proximal growth plate of the right humerus in a 13-year-old boy. B: Sessile variant. Broad-based osteochondroma is seen here arising from the medial cortex of the proximal diaphysis of the right humerus in a 14-year-old boy. Note that in both lesions the cortex of the host bone merges, without interruption, with the cortex of the lesion, and the medullary portion of both lesions and the medullary cavities of the adjacent bones communicate. |
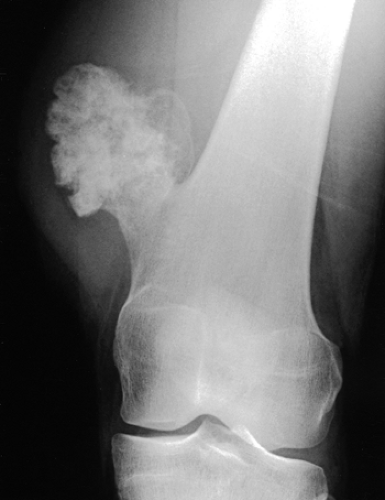 Figure 3-40 Osteochondroma. The lesion exhibits calcifications in the chondroosseous zone of the stalk. |
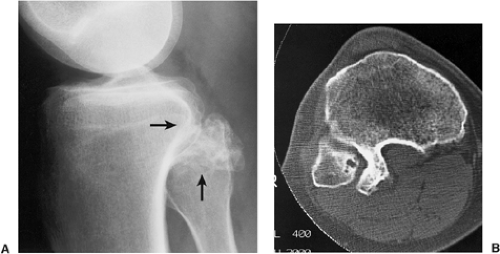 Figure 3-41 Osteochondroma: computed tomography (CT). A: Lateral radiograph of the knee shows a calcified lesion at the posterior aspect of the proximal tibia (arrows). The exact nature of this lesion can not be ascertained. B: CT clearly establishes the continuity of the cortex, which extends without interruption from the osteochondroma into the tibia. Note also that the medullary portion of the lesion and the tibia communicate. |
The Langer-Giedion syndrome, also known as trichorhinophalangeal syndrome type II, is characterized in addition to multiple osteochondromas by craniofacial dysmorphism and mental retardation. It is due to deletion of 8q24. Similar to the former disorder is Potocki-Shaffer syndrome, characterized by multiple osteochondromata, enlarged parietal foramina, and sometimes craniofacial dysostosis and mental retardation (80).
The pathologic features of multiple osteochondromata are the same as those of solitary lesions (99). Malignant transformation to chondrosarcoma [and in rare instances to other types of sarcoma (80)] in osteochondromatosis is more common than in solitary lesions, not only because of the greater number of lesions present but because of the higher risk for malignant transformation of each lesion. However, estimated figures from older literature (ranging from 5% to 25%) seem to be too high, mainly because they have been obtained from large tumor referral centers. Recent investigations point to a much lower percentage (1%–3%) of sarcomatous transformation of these lesions (111,114). Lesions at the shoulder girdle and around the pelvis are usually at greater risk for this complication. Malignant transformation before the age of 20 is uncommon.
As previously stated, hereditary osteochondromatosis is a heterogeneous autosomal dominant disorder in
which three gene loci have been identified on chromosomes 8q24.1 (EXT1), 11p11–p12 (EXT2), and 19p (EXT3). So far, EXT1 and EXT2 have been cloned. These loci encode glycoproteins that are involved in biosynthesis of heparan sulfate proteoglycans. Several mutations have been detected. On the basis of analogue findings in the fruit fly Drosophila melanogaster, it appears that alteration of heparan sulfate biosynthesis may interfere with the normal maturation of chondrocytes, leading to premature chondrocyte differentiation and subsequent ossification (80,85). Because genotype–phenotype studies have shown that patients with an EXT1 mutation, as compared to patients with an EXT2 alteration, are at increased risk for development of severe disease and sarcomas, screening for EXT1 mutations in these patients appears to be advisable (89,112).
which three gene loci have been identified on chromosomes 8q24.1 (EXT1), 11p11–p12 (EXT2), and 19p (EXT3). So far, EXT1 and EXT2 have been cloned. These loci encode glycoproteins that are involved in biosynthesis of heparan sulfate proteoglycans. Several mutations have been detected. On the basis of analogue findings in the fruit fly Drosophila melanogaster, it appears that alteration of heparan sulfate biosynthesis may interfere with the normal maturation of chondrocytes, leading to premature chondrocyte differentiation and subsequent ossification (80,85). Because genotype–phenotype studies have shown that patients with an EXT1 mutation, as compared to patients with an EXT2 alteration, are at increased risk for development of severe disease and sarcomas, screening for EXT1 mutations in these patients appears to be advisable (89,112).
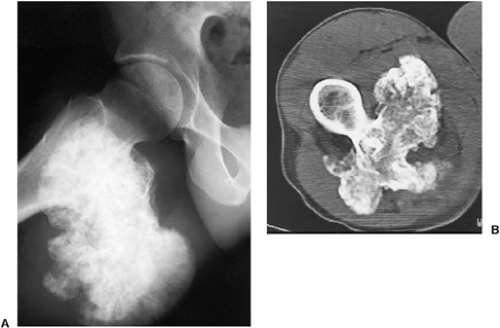 Figure 3-42 Osteochondroma: computed tomography (CT). A: Radiograph shows a large osteochondroma arising at the intertrochanteric area of the right femur. B: CT section shows the heavily calcified, cauliflower-like mass and a thin, cartilaginous cap. |
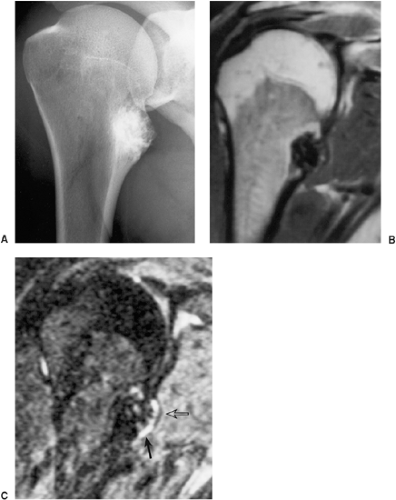 Figure 3-43 Osteochondroma: magnetic resonance imaging (MRI). A: Radiograph of the right shoulder shows a sessile lesion at the medial aspect of the proximal humeral diaphysis. B: T1-weighted coronal MRI reveals that the lesion exhibits a low signal intensity because of significant mineralization. C: T2-weighted image shows the thin, cartilaginous cap as an area of high signal intensity (arrow), covered by a linear area of low signal intensity representing perichondrium (open arrow). |
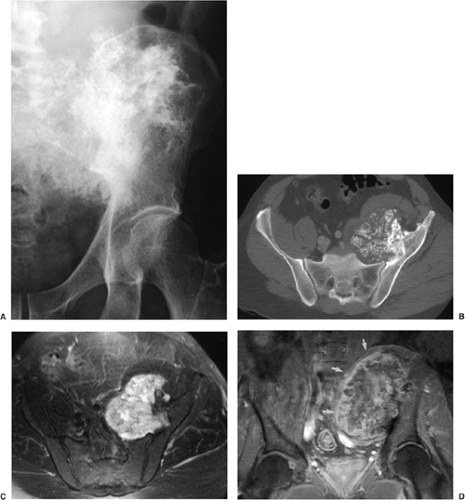 Figure 3-44 Osteochondroma: computed tomography (CT) and magnetic resonance imaging (MRI). A: Anteroposterior radiograph of the left hemipelvis of a 29-year-old man shows a large sessile osteochondroma arising from the ilium. B: Axial CT section demonstrates better the relationship between the lesion and the host bone. C: The cartilaginous cap of osteochondroma exhibits high signal intensity on axial T2-weighted MRI. D: After intravenous administration of gadolinium (Gd-DTPA) there is peripheral enhancement of the fibrovascular layer which covers the nonenhancing cartilaginous cap (arrows). |
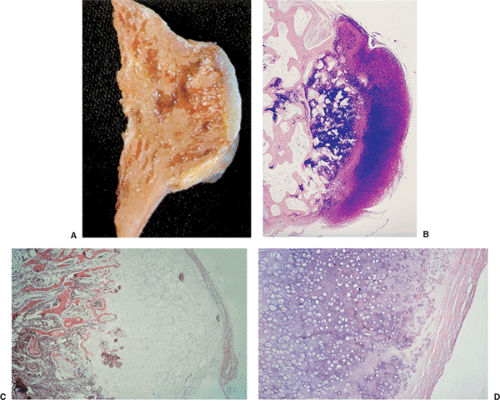 Figure 3-45 Histopathology of osteochondroma. A: Gross specimen of sessile osteochondroma exhibits characteristic features of the lesion: continuity of the cortex and medullary cavity and a thin, cartilaginous cap. (Reprinted with permission from Bullough PG. Atlas of orthopedic pathology, 2nd ed. New York: Gower, 1992:14.10.) B: The tumor consists of the hyaline cartilage cap with strong metachromasia of the matrix. A broad zone of endochondral ossification borders the cancellous bone containing the remnants of cartilaginous matrix (center) (Giemsa, original magnification ×6). C: At higher magnification areas of calcification are seen in the chondroosseous portion of the lesion. These areas correspond to a zone of provisional calcification in the growth plate (hematoxylin and eosin, original magnification ×30). D: Hyaline cartilage cap is seen at the periphery of the lesion, and beneath it transformation of cartilage into bone by endochondral ossification. A thin fibrous membrane (perichondrium) loosely overlies the cartilage (hematoxylin and eosin, original magnification ×60). |
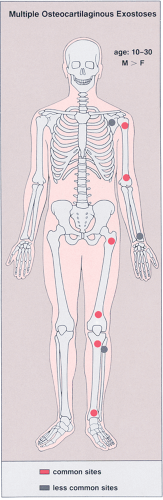 Figure 3-46 Multiple osteocartilaginous exostoses: skeletal sites of predilection, peak age range, and male-to-female ratio. |
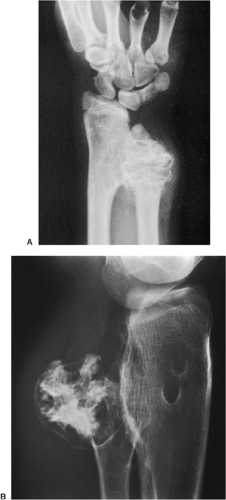 Figure 3-47 Multiple osteocartilaginous exostoses. A: Radiograph of the distal forearm of an 8-year-old boy with multiple osteochondromas shows growth disturbance in the distal radius and ulna. B: In another patient, a 21-year-old woman, observe growth disturbance of the proximal fibula. |
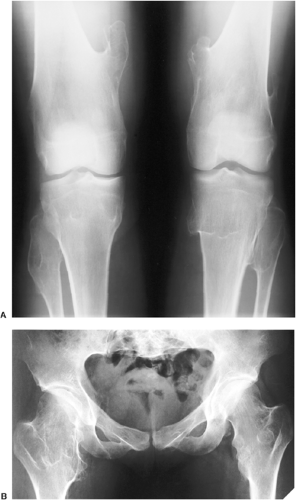 Figure 3-48 Multiple osteocartilaginous exostoses. A: Anteroposterior radiograph of both knees of a 17-year-old boy shows numerous sessile and pedunculated lesions. B: Anteroposterior radiograph of the hips shows numerous sessile osteochondromas affecting proximal femora. |
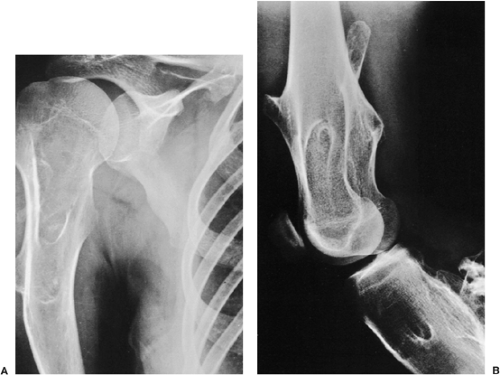 Figure 3-49 Multiple osteocartilaginous exostoses. A: Anteroposterior radiograph of the shoulder of a 22-year-old man with familial multiple osteochondromas demonstrates multiple sessile lesions involving the proximal humerus and scapula. B: Involvement of the distal femur and proximal tibia in the same patient is characteristic of this disorder. |
Differential Diagnosis
Radiology
The most important differential diagnosis is the distinction between benign osteochondroma and exostotic chondrosarcoma arising in previous exostosis. It is important to recognize early the features that suggest malignant transformation of osteochondroma to chondrosarcoma. Radiologic evaluation may reveal a constellation of features suggesting this complication: development of a thick, bulky cartilaginous cap (usually more than 2 to 3 cm in thickness) (95,105); development of a soft tissue mass with or without calcifications; and dispersed calcifications within the cartilaginous cap, separate from those contained in the stalk (Fig. 3-52), a sign described by Norman and Sissons (110). The most reliable imaging modalities for evaluating possible malignant transformation are radiography, CT, and MRI (83,150). Radiographs usually demonstrate containment of the calcifications of osteochondroma within the stalk of the lesion, but occasionally conventional tomography can also be helpful (Fig. 3-53). Dispersement of the calcifications in the cartilaginous cap and increased thickness of the cap, the cardinal signs of malignant degeneration, can be demonstrated by any of the previously mentioned modalities. Radionuclide bone scan can also be performed and may reveal increased uptake of radiopharmaceutical agent at the site of a lesion. Exostotic chondrosarcoma often exhibits greater intensity of uptake than a benign exostosis. However, a number of investigators believe that this is not always a reliable distinguishing feature of malignant transformation (94). The increased activity of a benign exostosis on bone scan is related to endochondral ossification within the cartilaginous cap, whereas that of exostotic chondrosarcoma represents active ossification, osteoblastic activity, and hyperemia within the cartilage and bony stalk of the
tumor (94). Occasionally the clinical and radiologic features of inflammatory changes occurring in bursa exostotica that covers the cartilaginous cap of osteochondroma can mimic malignant transformation to chondrosarcoma (Figs. 3-54 and 3-55).
tumor (94). Occasionally the clinical and radiologic features of inflammatory changes occurring in bursa exostotica that covers the cartilaginous cap of osteochondroma can mimic malignant transformation to chondrosarcoma (Figs. 3-54 and 3-55).
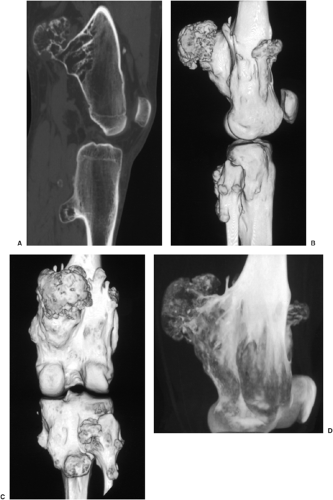 Figure 3-50 Multiple osteocartilaginous exostoses: three-dimensional computed tomography (3-D CT). A: Conventional sagittal reformatted CT section shows osteochondromas arising from the posterior aspects of distal femur and proximal tibia. B, C: Three-dimensional CT images viewed from lateral and posterior aspects of the knee show spatial distribution of numerous osteochondromas. D: 3-D CT image of the distal femur in maximum intensity projection (MIP) shows interior architecture of one of the sessile osteochondromas. |
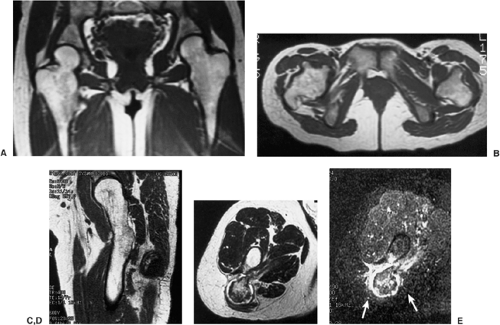 Figure 3-51 Multiple osteocartilaginous exostoses: magnetic resonance imaging (MRI). Coronal (A) and axial (B) T1-weighted (SE, TR 600, TE 20) MR images show several sessile osteochondromas affecting the proximal femora. Observe abnormal tubulation of the bones. C: In another patient with multiple cartilaginous exostoses MRI was performed because one of the osteochondromas continued to enlarge. Sagittal T1-weighted (SE, TR 400, TE 12) image demonstrates intact cortex covering the lesion. D: Axial fast-spin echo (FSE, TR 4000, TE 102 Ef) MRI shows high-intensity thin cartilaginous cap of osteochondroma without malignant changes. E: Lack of malignant transformation was confirmed on axial inversion recovery (FMPIR/90; TR 4000, TE 51 Ef, TI 140) MRI, which shows no soft tissue extension of the lesion (arrows). |
On radiographic examination, the signs pointing to malignant transformation of multiple cartilaginous exostoses to chondrosarcoma are identical with those present in malignant transformation of solitary osteochondroma (90). After skeletal maturation has occurred, alteration in a lesion’s size must be considered a potential indicator of malignant transformation. Particularly suggestive are alterations in the size and irregularity of contour of the cartilage cap, or the presence of mineral deposition beyond the previous contour as documented by radiography (18). When such signs are associated with signs of bone destruction at the base or neck, or when a soft tissue mass is present, the diagnosis of malignancy is certain (Fig. 3-56).
An interesting lesion that can be confused with osteochondroma is the so-called epiphyseal or intraarticular osteochondroma, better known as Trevor-Fairbank disease or dysplasia epiphysealis hemimelica (89,98). This is a developmental disorder characterized by asymmetric cartilaginous overgrowth of one or more epiphyses in the lower and occasionally the upper extremities. The talus, distal femur, and distal tibia are the most common sites of involvement (98). The lesion typically arises on one side of the affected limb and deforms the bone. Males are affected approximately three times as often as females. The basic pathologic process is abnormal cartilage proliferation in the epiphysis, and on histologic examination the lesion is almost identical with osteochondroma. Dysplasia epiphysealis hemimelica has been noted in association with conventional osteochondroma, chondroma (93), and Ollier disease (58). The clinical features are pain, deformity, and restricted motion in the affected joints. Radiography reveals an irregular, bulbous overgrowth of the epiphysis to one side of the ossification center (Fig. 3-57). Although CT may demonstrate the continuity of the mass with the underlying epiphysis, MRI is more effective in assessment of this abnormality. As the lesion enlarges the joint deformity increases (100).
Finally, sessile osteochondroma should be differentiated from periosteal chondroma (26). The latter is separated
from the host bone by intervening cortex (see also section on differential diagnosis of periosteal chondroma). Differentiation of sessile osteochondroma from occasionally similarly appearing parosteal osteosarcoma, soft tissue osteosarcoma, and myositis ossificans should not create a significant problem (Fig. 3-58).
from the host bone by intervening cortex (see also section on differential diagnosis of periosteal chondroma). Differentiation of sessile osteochondroma from occasionally similarly appearing parosteal osteosarcoma, soft tissue osteosarcoma, and myositis ossificans should not create a significant problem (Fig. 3-58).
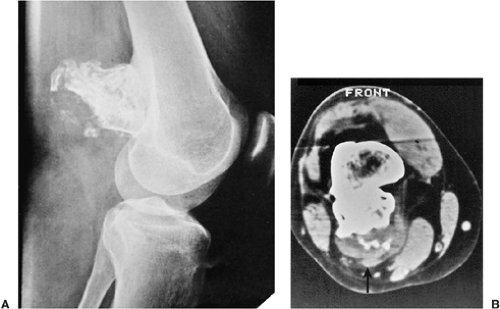 Figure 3-52 Exostotic chondrosarcoma. A 28-year-old man developed pain in the popliteal region and also noted an increase in a mass of which he had been aware for 15 years. A: Lateral radiograph of the knee shows a sessile-type osteochondroma arising from the posterior cortex of the distal femur. Calcifications are present not only in the stalk of the lesion but are also dispersed in the cartilaginous cap. B: Computed tomography section confirms the increased thickness of the cartilaginous cap (2.5 cm) and dispersed calcifications within the cap (arrow). These features are consistent with a diagnosis of malignant transformation to chondrosarcoma, which was confirmed by histopathologic examination. |
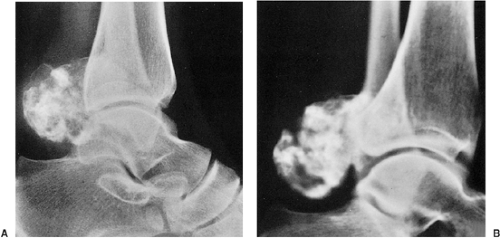 Figure 3-53 Osteochondroma resembling exostotic chondrosarcoma. A: Lateral radiograph of the ankle of a 26-year-old woman with a painful osteochondroma shows a sessile lesion arising from the posterior aspect of the distal tibia. In the interpretation of this radiograph, uncertainties were raised that some of the calcifications might not be contained within the stalk, and tomographic examination was suggested. B: Tomographic section demonstrates lack of separation of calcifications from the main mass, suggesting a benign lesion. The osteochondroma was resected and histopathologic examination confirmed the lack of malignant transformation. |
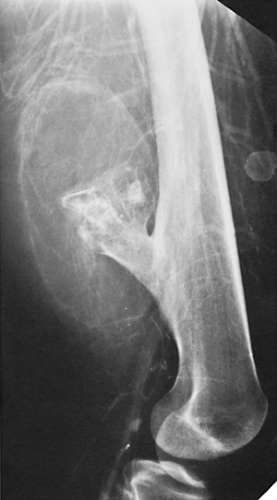 Figure 3-54 Bursa exostotica. A 25-year-old man with a known solitary osteochondroma of the distal right femur presented with gradually increasing pain. Malignancy was suspected and arteriography was performed. The capillary phase of the arteriogram reveals a huge bursa exostotica. Inflammation of the bursa, with accumulation of a large amount of fluid (bursitis), was the cause of the patient’s symptoms. |
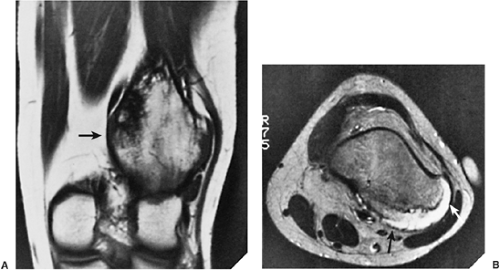 Figure 3-55 Bursa exostotica. A 12-year-old girl presented with pain in the popliteal fossa. A: Coronal T1-weighted (SE, TR 650, TE 25) magnetic resonance imaging (MRI) demonstrates a large osteochondroma arising from the posterolateral aspect of the distal femur (arrow). B: Axial T2-weighted (SE, TR 2200, TE 70) MRI shows a bursa exostotica distended with high-intensity fluid (arrows). |
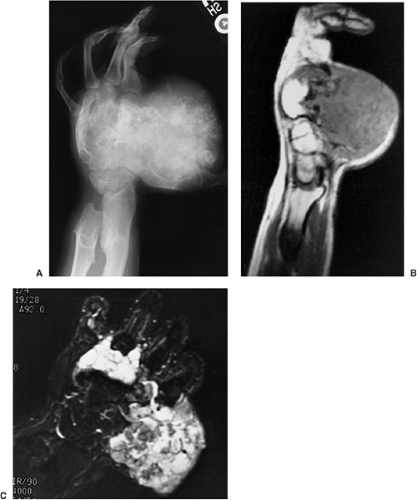 Figure 3-56 Multiple cartilaginous exostoses: malignant transformation to chondrosarcoma. A: Oblique radiograph of the hand of a 22-year-old man shows multiple osteochondromas. A large soft tissue mass with chondroid calcifications indicates malignant transformation. B: Sagittal T1-weighted (SE, TR 600, TE 16) magnetic resonance imaging (MRI) reveals volar extension of a large soft tissue mass. C: Coronal inversion recovery (FMPIR/90, TR 4000, TE 64/Ef) MRI demonstrates malignant lobules of the cartilage invading the bones and soft tissues of the hand. |
Pathology
Malignant transformation of osteochondroma to chondrosarcoma is the most important histopathologic differential diagnosis. On histologic examination, malignant transformation is recognized by greater cellularity of the cartilage tissue, uneven distribution of cells without cord- or column-like arrangement, pleomorphism, and cellular and nuclear atypia (Fig. 3-59). Additional criteria include destruction of bone trabeculae of the stalk with invasion of marrow spaces, and invasion of the overlying soft tissues.
The other differential possibility of osteochondroma is so-called reactive cartilaginous exostosis, which differs from the former by its smaller size and the metaplastic formation of cartilage from the fibrous tissue covering a bone (108) (Fig. 3-60). The resulting fibrocartilage undergoes endochondral ossification, as
in osteochondroma. Continuing formation is the result of ongoing metaplasia from the surrounding fibrous tissue on its surface, whereas in osteochondroma growth is generated by mitotic division of the chondrocytes.
in osteochondroma. Continuing formation is the result of ongoing metaplasia from the surrounding fibrous tissue on its surface, whereas in osteochondroma growth is generated by mitotic division of the chondrocytes.
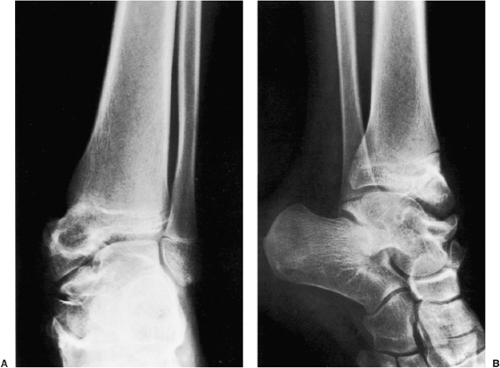 Figure 3-57 Dysplasia epiphysealis hemimelica. A 12-year-old girl presented with pain and limitation of motion in the ankle joint. Anteroposterior (A) and lateral (B) radiographs of the ankle demonstrate deformity and enlargement of the medial malleolus, talus, and navicular bone, features typical of Trevor-Fairbank disease. |
Bizarre parosteal osteochondromatous proliferation (BPOB or Nora lesion) can be excluded by its feature of not being in continuity with the marrow cavity of underlying bone. Furthermore, Nora lesion is characterized by a spindle-cell stroma, irregular ossifications, and cap-like irregularly structured cartilage; however, cellular atypia is absent (82,106,109) (see Fig. 2-131C).
An osteochondroma in which intracartilaginous ossification has completely consumed the cartilage may be mistaken for an osteoma if the base of the stem is not available for histologic examination.
Chondroblastoma (Codman Tumor)
Clinical Presentation
Representing less than 1% of all primary bone tumors and 9% of benign bone tumors, this rare lesion typically presents in the epiphysis of long bones, such as the humerus, tibia, and femur (121,177,178). So far, only one case of diaphyseal chondroblastoma in a long bone has been reported (118a). The proximal humerus, distal femur, and proximal tibia are the preferred sites of involvement (153) (Fig. 3-61). Occasionally the patella, which is considered equivalent to an epiphysis, is involved (164). Ten percent of chondroblastomas involve the small bones of the hands and feet, the talus and calcaneus representing the most common sites (125,130,154,158,166). Only 1% of all chondroblastomas have been reported to involve the skull (152). Involvement of the vertebrae is exceedingly rare (144,157). The lesion is usually seen before skeletal maturity, but some cases have been reported after obliteration of the growth plate (129). Males are almost twice as frequently affected as females (52), although in the series from the Armed Forces Institute of Pathology (AFIP) the ratio was 3:1 (164). In general, the clinical symptoms are nonspecific: pain, often related to the nearest joint, and swelling, usually lasting for several months, are the most common complaints (123,132). Approximately one third of patients exhibit joint effusion (174). Pathologic fractures occur only rarely.
Imaging
Radiographically, the lesion is usually located eccentrically (168). It is radiolucent and well defined, usually with a thin sclerotic border, and it exhibits a geographic pattern of bone destruction (141) (Fig. 3-62).
The margins are smooth in 63% of cases, scalloped in 27%, and lobulated in 10% (161). Internal trabeculation can sometimes be observed (156). In 25% to 50% of cases, stippled calcifications are present (Fig. 3-63). If calcifications are not apparent on standard radiographs, conventional tomography or CT can be helpful (138) (Fig. 3-64; see also Fig. 3-62C). The latter modality may also assess the extent of the lesion (170). The presence of a solid or layered periosteal reaction (123), usually located in some distance from the tumor, was reported on conventional radiographs in more than 50% of 214 cases in the series by Brower et al. (126) (Fig. 3-64A; see also Fig. 3-65A). These authors suggest an inflammatory response to the tumor as the cause of this phenomenon. MRI usually reveals a larger area of involvement than can be seen on radiography, including prominent soft tissue and regional bone marrow edema (128,139,181) (Fig. 3-65). This strikingly aggressive appearance can lead to an incorrect diagnosis of a malignant tumor (155,180). The combination of highly cellular chondroid matrix and calcification in chondroblastoma presumably accounts for the observed decrease in relaxation time on T2-weighted imaging (128). Low to intermediate heterogeneous signal intensity, lobular internal architecture, and fine lobular margins were detected with high-resolution T2-weighted MRI (180). Focal lobules of low, intermediate, and high signal intensity are superimposed and most likely correspond to calcification, chondroid matrix, and fluid in the lesion. In addition, MRI may demonstrate a rare extension of tumor to the soft tissues and may also reveal a joint effusion (Fig. 3-66).
The margins are smooth in 63% of cases, scalloped in 27%, and lobulated in 10% (161). Internal trabeculation can sometimes be observed (156). In 25% to 50% of cases, stippled calcifications are present (Fig. 3-63). If calcifications are not apparent on standard radiographs, conventional tomography or CT can be helpful (138) (Fig. 3-64; see also Fig. 3-62C). The latter modality may also assess the extent of the lesion (170). The presence of a solid or layered periosteal reaction (123), usually located in some distance from the tumor, was reported on conventional radiographs in more than 50% of 214 cases in the series by Brower et al. (126) (Fig. 3-64A; see also Fig. 3-65A). These authors suggest an inflammatory response to the tumor as the cause of this phenomenon. MRI usually reveals a larger area of involvement than can be seen on radiography, including prominent soft tissue and regional bone marrow edema (128,139,181) (Fig. 3-65). This strikingly aggressive appearance can lead to an incorrect diagnosis of a malignant tumor (155,180). The combination of highly cellular chondroid matrix and calcification in chondroblastoma presumably accounts for the observed decrease in relaxation time on T2-weighted imaging (128). Low to intermediate heterogeneous signal intensity, lobular internal architecture, and fine lobular margins were detected with high-resolution T2-weighted MRI (180). Focal lobules of low, intermediate, and high signal intensity are superimposed and most likely correspond to calcification, chondroid matrix, and fluid in the lesion. In addition, MRI may demonstrate a rare extension of tumor to the soft tissues and may also reveal a joint effusion (Fig. 3-66).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


