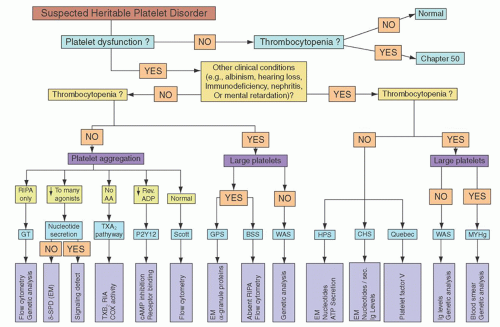
The preliminary diagnosis of platelet dysfunction must be made on the basis of patient history, and the diagnosis then confirmed by specific laboratory tests of platelet function. A practical algorithm for this purpose, depicted in
Figure 52.5, is reproduced with permission from the excellent review by Bolton-Maggs et al.
5
Bedside Exam and Patient History
An accurate and detailed patient and family history is a key element in the assessment of platelet disorders. One should bear in mind that bleeding histories are subjective and that bleeding can be variable, often evolving or decreasing throughout a person’s lifetime. In particular, children may not have yet had enough hemostatic challenges to develop a strong clinical. The subjectivity of the oral history is reflected in a recent statistic that at least one quarter of persons who complain of serious bleeding do not have a bleeding disorder, whereas at least one third of persons who
have no bleeding can be shown to have von Willebrand disease (vWD) or a platelet disorder.
6To overcome the subjectivity of a history for superficial mucocutaneous bleeding, the International Society on Thrombosis and Hemostasis (ISTH) suggests that bleeding should be considered clinically significant when there are two or more distinct bleeding sites such as the skin, nose, gums, vagina, gastrointestinal tract, or genitourinary tract. This includes either spontaneous bleeding or provoked bleeding, such as that which might result from dental work, parturition, trauma, or surgery. In addition, a bleeding history involving only a single site should be considered significant when it is so severe as to warrant blood transfusions. Finally, a single bleeding symptom that recurs on three or more unrelated and separate occasions should also be considered significant.
7,
8A number of quantitative approaches to assess the relative severity of patient bleeding have been proposed, some including a normalization based on patient age.
9,
10 However, such approaches work best for a comparison of cohorts of related individuals with comparable bleeding disorders, such as families of patients with vWD.
The distinction between normal individuals and those with bleeding disorders is not always clear cut. Care should be taken in all patients to note the use of medicines, either “over the counter,” herbal, or prescription, that are known to influence platelet function. This is particularly true of medications such as aspirin, other nonsteroidal anti-inflammatory drugs (NSAIDs), ticlopidine or clopidogrel, integrin
αIIb
β3 antagonists (e.g., Abciximab, tirofiban, and eptifibatide), epoprostenol, statins, cilostazol, sildenafil, fluoxetine, and large doses of various
β-lactam antibiotics (penicillins > cephalosporins).
7Bleeding manifestations typical of platelet dysfunction include: (1) Unexplained or extensive bruising; (2) epistaxis, particularly if lasting more than 30 minutes, causing anemia or admission to hospital; (3) menorrhagia, particularly if this has been present since the menarche; (4) oral cavity bleeding; (5) bleeding during
childbirth; (6) bleeding following invasive procedures; and (7) bleeding following dental extraction.
Severe platelet dysfunction is present from early childhood onward. Following delivery of an affected infant one may find intracranial and/or subdural hemorrhage, excessive bleeding from the umbilical stump or after circumcision, or easy bruising after handling. As the infant becomes more mobile, easy or extensive bruising following relatively mild trauma can be indicative of platelet dysfunction. Prolonged epistaxis is a common finding throughout childhood and can even become life threatening. In adults, menorrhagia and bleeding during childbirth are common and potentially serious, whereas bleeding following any invasive procedure should be anticipated.
5Mild platelet dysfunction is more likely to manifest itself at any age most commonly following a definable hemostatic challenge, such as surgery or dental extractions. Easy bruising is a very nonspecific symptom, and many cases are difficult to distinguish from what would otherwise be considered a normal response.
Consanguinity increases the likelihood of an autosomal recessive platelet disorder, and a family history is invaluable in establishing the diagnosis of inherited platelet dysfunction.
Laboratory Assessment
A number of laboratory-based evaluations are critical for an accurate diagnosis of platelet dysfunction.
Whole Blood Platelet Count
A key element in assessing platelet dysfunction is an accurate whole blood platelet count. Automated counts should be viewed as provisional. Inasmuch as macrothrombocytes or platelet aggregates created inadvertently during blood processing will not be counted, the whole blood platelet count should be confirmed by an optical method.
Global Coagulation Tests
To rule out abnormalities in clotting factors, all patients should have a prothrombin time, activated partial thromboplastin time, and thrombin time performed. Laboratories should determine their own age-related normal range. It is also critical to investigate all patients for VWD, which is far more common than platelet function disorders and creates a similar bleeding phenotype.
Bleeding Time
The bleeding time is historically a common measure of platelet function. However, because the test is poorly reproducible, time consuming, and insensitive, the bleeding time has gradually fallen out of favor as a clinically useful test.
11 In cases of mild platelet dysfunction, the bleeding time is often normal or minimally prolonged;
11 in severe cases, it will usually be prolonged.
Unfortunately, the bleeding time does not correlate well with the in vivo bleeding tendency within individual patients, and an accurate bleeding history is considered by many to be a more valuable screening test. Nonetheless, a prolonged bleeding time should be considered sufficient grounds to perform additional tests of platelet function.
Platelet Function Analyzer-100
The platelet function analyzer-100 (PFA-100) measures the rate of thrombus formation under high shear in citrated whole blood that is perfused through a membrane aperture coated with collagen/epinephrine or collagen/ADP. The closure time (CT) will be significantly prolonged in Glanzmann thrombasthenia (GT) and Bernard-Soulier syndrome (BSS) using either ADP/collagen or epinephrine/collagen membrane cartridges.
12,
13 Consequently, the PFA-100 can be used to screen patients to exclude these diagnoses.
The PFA-100 may be sensitive to platelet storage pool disease (SPD), primary secretion defects, the Hermansky-Pudlak syndrome (HPS), and the Quebec syndrome. However, because falsenegative results occur in patients with all of these disorders, there are those who question its usefulness as a screening tool.
12,
14 The PFA-100 is affected by platelet count and hematocrit, and is dependent on normal VWF levels and naturally occurring (genetic) differences in platelet membrane GPVI or
α2
β1 expression.
13,
14,
15
Platelet Aggregation
Platelet aggregation in platelet-rich plasma remains an important test in the analysis of platelet function. The typical agonists that are used to induce platelet aggregation are ADP, epinephrine, collagen, arachidonic acid (AA), ristocetin, the TX receptor agonist U46619, thrombin or the thrombin receptor-activating peptide. Because the level and/or activity of each of the receptors for the agonists can vary among normal subjects, it is recommended that dose-response curves to each agonist be obtained from the patient under study and compared to a reference range obtained from multiple normal subjects.
16 When thrombin is the agonist, an inhibitor of fibrin polymerization, such as glycine-proline-arginine-proline peptide, must be added to the PRP, or alternatively, plasma-depleted washed platelets must be used.
Platelet aggregation is sensitive to platelet count, and at counts ≤120,000/µl, the response to some agonists will be impaired. In thrombocytopenic samples, the best options are to adjust a control sample to the same count as the patient or to perform studies on washed platelets where the platelet number can be normalized. Consideration should also be given to more specialized tests, such as a measure of P-selectin expression or integrin αIIbβ3 activation by flow cytometry. The expected aggregation responses associated with specific diagnoses are covered in the appropriate sections.
Adenine Nucleotide Content and Release
Measurement of platelet adenosine nucleotide (ADP and adenosine triphosphate [ATP]) content and release can be used in the diagnosis of storage pool and release defects.
17 A finding of normal platelet aggregation does not exclude the diagnosis of SPD. It is recommended that patients suspected of having platelet dysfunction should have both platelet aggregation and adenine nucleotide release performed, unless it is certain that the laboratory performing the platelet aggregation assays can demonstrate that their assay conditions are sensitive to defects in platelet nucleotide amount or release.
Platelet nucleotide content and release varies with age. Ideally, age-related normal ranges should be established for total and released levels of ATP and ADP and their ratios. Platelet aggregation, nucleotide content, and nucleotide release in children over 12 months of age do not differ significantly from adult values, whereas collagen-induced platelet nucleotide release has been shown to be reduced in neonates compared with children older than 1 year. Agonist-induced secretion of platelet granule contents has been shown to be reduced in both term and premature babies due to immature signal transduction pathways.
18,
19
Flow Cytometry
Flow cytometry is routinely used to measure platelet surface receptor density, platelet activation,
α-granule release, procoagulant phospholipid expression, and microvesicle production.
20,
21 A common application of flow cytometry is the assessment of GPIb complex and integrin
αIIb
β3 expression in the diagnosis of BSS and GT. Individuals who are heterozygous for these disorders are also readily distinguished. An important benefit of flow cytometry is the small quantities of blood required, an attractive feature in young children or thrombocytopenic individuals.
Electron Microscopy
Transmission electron microscopy (TEM) of fixed/embedded platelet thin sections can be performed by a limited number of specialized personnel, but is critical in the assessment of platelet granule defects and changes in platelet ultrastructure (e.g., in the evaluation of patients with
MYH-9 defects). Whole-mount EM can be used to quantitate
δ-granule content because this is readily identified in unstained preparations.
22
Additional Assays
Several additional assays are available in specialized laboratories that can provide further information relevant to the diagnosis of the particular platelet disorder, including analysis of receptor expression, specific molecular or genetic defects, protein phosphorylation, formation of signal transduction intermediates, or a characterization of the platelet proteome. The utility of these tests in clinical diagnoses is currently under intense investigation.
Overview of Treatment Options for Platelet Disorders
Although the number of clearly identifiable platelet dysfunction syndromes is growing, our choice of treatment options remains limited. Minor membrane bleeding may be controlled with topical agents in the nasal or oral cavities with antifibrinolytic agents. Epsilon-aminocaproic or tranexamic acids will decrease blood loss associated with epistaxis or menorrhagia. Some patients may benefit from Stimate as documented by DiMichele and Hathaway,
23 whereas others actually bleed more with this agent due to the fibrinolysis induced by this medication. For this reason many centers recommend the use of Stimate in conjunction with Amicar or Cyklokapron.
In the case of menorrhagia, hormonal suppression is the mainstay for women who do not wish to undergo endometrial ablation for hysterectomies. In combination with antifibrinolytics and Stimate, even severe bleeding may be controlled; however, more aggressive therapy including platelet infusion may be required to control bleeding before these agents take effect.
Platelet transfusion may pose a dilemma to those physicians wishing to avoid alloimmunization in patients who may require multiple transfusions throughout their lifetimes to control hemorrhage. This is particularly true for those patients who are lacking membrane GPs, such as αIIbβ3 or GPIb. In this setting patients are at risk of developing isoimmunization, making antibodies against the “foreign” proteins that they lack and thus becoming refractory to all subsequent platelet transfusions.
Although rare, this creates a significant challenge in patients who require frequent platelet infusions to control life-threatening bleeding. Patients with GT have not only developed antibodies to αIIb and/or β3 but also demonstrated anti-idiotypic antibodies that bind to fibrinogen, thus creating a hemorrhagic disorder far worse than the underlying platelet dysfunction. In general, physicians avoid platelet transfusion apart from cases of severe hemorrhage. Isoimmunization appears to be rare in Glanzmann but the risk of alloimmunization is still a major concern, therefore leukocyte depletion of transfused platelets is recommended to decrease the frequency of sensitization.
Activated recombinant Factor VII (rFVIIa) has been used to slow or arrest bleeding associated with platelet dysfunction.
24 Dosages have varied widely but many patients have responded to this regimen when others have failed. Used in combination with antifibrinolytics, minor bleeding can be controlled in certain patients. This treatment is often used prior to platelet transfusion in order to avoid blood product exposure and isoimmunization.
For those patients who present with recurrent life-threatening bleeds, bone marrow transplant or stem cell infusion following immune ablation is recommended before the patients have extensive blood product exposure.
25 Successful transplantation with normal stem cells represents long-term cure for these patients. Although complications related to stem cell transplantation cannot be overlooked, successful engraftment essentially eliminates the significant risk of mortality related to hemorrhage in patients with severe disorders.
Patients with rare platelet dysfunction syndromes are now included in many of the rare bleeding disorder international and regional registries that will aid physicians in understanding the natural course, optimal therapy, and life expectancy for each of the syndromes listed below, as well as the numerous platelet disorders that have yet to be defined. Definitive molecular and biochemical diagnoses will dictate appropriate therapy in these patients. With improvement in platelet function measurement and proteomic approaches one should see significant improvement in early diagnosis and medical management.
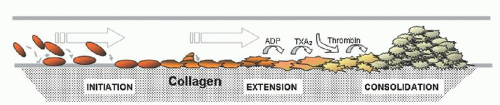
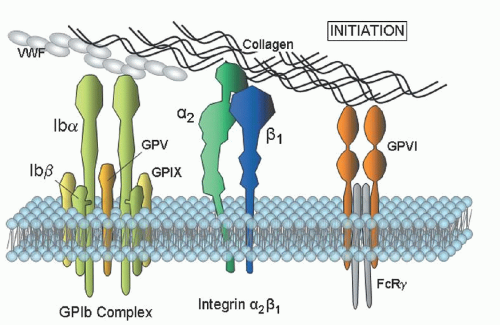
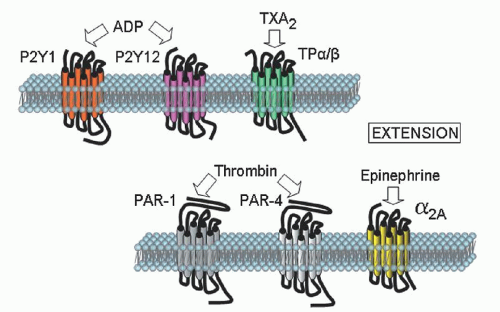
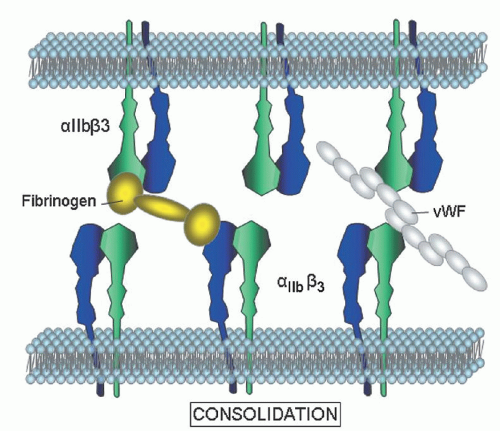
 Clinical Flow Cytometry
Clinical Flow Cytometry



