INTRODUCTION
SUMMARY
Pure red cell aplasia is the diagnosis applied to isolated anemia secondary to failure of erythropoiesis. Cardinal findings are a low hemoglobin level combined with reticulocytopenia and absent or extremely infrequent marrow erythroid precursor. Historical names for pure red cell aplasia include erythroblast hypoplasia, erythroblastopenia, red cell agenesis, hypoplastic anemia, and aregenerative anemia. Aplastic anemia confers the same meaning, of course, but is applied to pancytopenia and an empty marrow (Chap. 35). Pure red cell aplasia was first separated from aplastic anemia by Kaznelson in 1922. The association of red cell aplasia and thymoma interested physicians in the 1930s and ultimately led to laboratory studies linking pure red cell aplasia to immune mechanisms, including the early identification of antierythroid precursor cell antibodies by Krantz and later characterization of T cells that inhibited erythropoiesis. Red cell aplasia as an acute and life-threatening complication of sickle cell disease and other hemolytic anemias was recognized in the 1940s, presaging the role of a specific virus in the etiology of both acute and chronic erythropoietic failure. Despite its infrequency, pure red cell aplasia has been a subject of much laboratory research because of its link to an immune mechanism of erythropoietic failure and as a manifestation of parvovirus B19 infection and viral destruction of red cell progenitors. However, because of its infrequency, pure red cell aplasia has not been the subject of large or controlled clinical trials; as a result, therapeutic recommendations are based on single cases or small series. Table 36–1 lists a practical classification of pure red cell aplasia.
Acronyms and Abbreviations:
B19, primate erythroparvovirus 1; BFU-E, burst-forming unit–erythroid; CD20, a cluster of differentiation molecule expressed on the surface of all mature B cells; CFU-E, colony-forming unit–erythroid; CLL, chronic lymphocytic leukemia; FA, Fanconi anemia; GATA1 gene, globin transcription factor 1; HLA, human leukocyte antigen; Ig, immunoglobulin; IL, interleukin; LGL, large granular lymphocytic leukemia; RPS14 and RPS19 genes, ribosomal protein S14 and S19 genes; STAT3 gene, signal transducer and activator of transcription 3 gene; T cell, thymus-derived lymphocyte.
INHERITED PURE RED CELL APLASIA (DIAMOND-BLACKFAN ANEMIA)
Anemia in infancy and early childhood associated with absent reticulocytes in the blood and erythroid precursor cells in the marrow was described by Joseph1 in 1936 as a “failure of erythropoiesis” and by Diamond and Blackfan2 in 1938 as “congenital hypoplastic anemia.” Gasser3 first reported a response of a patient to glucocorticoids in 1951, and Diamond and associates4 presented a series of treated patients. Genetic linkage studies have identified etiologic mutations in ribosomal protein genes.5,6,7,8,9 Hundreds of cases have been reported, and many excellent reviews have been published.10 Although Joseph was the first to describe the disorder, the anemia invariably is referred to as either Blackfan-Diamond or Diamond-Blackfan anemia.
An annual incidence of 5 cases per 1 million livebirths has been estimated from registry data.11 Well-characterized pedigrees are consistent with an autosomal dominant or, less often, recessive inheritance pattern. Sporadic cases are seen most frequently. Retrospective studies may reveal subtle hematologic or biochemical lesions, or an abnormal gene, in an affected parent or another relative without clinical anemia.12
Recent genetic studies have led to the characterization of Diamond-Blackfan anemia as a disease of ribosomal biogenesis.5,6,7,13,14 Linkage analyses of several dozen European families mapped to a site on chromosome 19q1315 and the finding of a translocation in one individual allowed cloning of the ribosomal protein S19 (RPS19) gene, which encodes a protein involved in ribosome assembly.5,6,7,8,9 Most mutations are whole gene deletions, translocations or truncations; this pattern suggests a mechanism of haploinsufficiency, and RPS19 behaves as a dominant gene.16 (Disruption of both copies of the gene in the mouse prevents implantation.17) RPS19 mutations occur in approximately 25 percent of patients with inherited red cell aplasia16,18; and mutations subsequently have been identified in multiple other ribosomal biogenesis genes (RPS10, RPS26 particularly) in other cases.16,19,20 More recently, a globin transcription factor 1 (GATA1) gene mutation was identified in a Diamond-Blackfan kindred, implicating a signal transduction pathway of erythroid differentiation as also causative of the syndrome.21 Experiments have implicated RPS14 in one of the myelodysplastic syndromes characterized by loss of 5q.22
Precisely how defects in ribosomal protein genes cause constitutional red cell aplasia is uncertain. Historically, Diamond-Blackfan anemia has been characterized by diminished erythroid progenitor cell numbers (colony forming unit–erythroid [CFU-E] and burst-forming unit–erythroid [BFU-E]).23,24 In cell culture, early, erythropoietin-independent erythropoiesis is relatively normal; the major defect is in the late stage of erythropoietin-dependent erythroid cell expansion and maturation.25 A defect in late erythroid differentiation is compatible with the classic findings of macrocytosis and increased hemoglobin F expression in inherited red cell aplasia. Granulopoiesis in the colony-forming unit–granulocyte-macrophage assay and the earlier hematopoietic progenitors as measured in vitro by long-term culture-initiating cell assay (an assay for an early multipotential hematopoietic progenitor) frequently are abnormal, but to a lesser degree than are CFU-E and BFU-E.26 In a zebrafish model, deficiency of rps19 in early embryogenesis caused a decrease in erythrocytes and also physical anomalies.27 In tissue culture experiments, silencing of RPS19 profoundly affects erythropoietic differentiation and, to lesser degrees, myelopoiesis.28,29 Both in vivo and in vitro models have implicated accumulation in the cell of free ribosomal proteins, which modulates the inhibitory activity of regulators of tumor protein p53, leading to p53 stabilization and apoptosis.30 The specificity of this molecular defect for the erythroid pathway may be a result of the extreme requirement of red cell progenitors and precursors for ribosome biogenesis.
Despite responsiveness of patients to glucocorticoids, there is little evidence of an immune mechanism, cellular or humoral, underlying inherited red cell aplasia.
Approximately one-third of patients are diagnosed at birth or within a few weeks of delivery, and almost all are identified within the first year of life.31 Considerable variations are noted with regard to severity of phenotype, ranging from hydrops fetalis32,33 to presentation in adulthood, when diagnosis is inferred from associated physical anomalies.5,34 No sex predominance exists. Increased rates of prematurity in patients and of miscarriages in families have been inferred from collected cases.35 Symptoms of anemia in early childhood include pallor, apathy, poor appetite, and “failure to thrive.” Physical anomalies occur in about a third of cases; most frequent is craniofacial dysmorphism, the classic appearance described by Cathie36 is “tow-colored hair, snub nose, wide set eyes, thick upper lips, and an intelligent expression.” Malformations of the thumbs and short stature are frequent, followed by abnormalities of the urogenital system, web neck, and skeletal and cardiac defects.11,31,37 These physical anomalies are less prevalent than the abnormalities seen in Fanconi anemia (FA).
The degree of anemia is highly variable at diagnosis. Erythrocytes may be macrocytic or normocytic. Reticulocytopenia is profound. The marrow, which usually is devoid of red blood cell precursors, may show small numbers of megaloblastoid early erythroid cells with apparent “maturation arrest.” Platelets are normal or elevated. Leukocytes may be normal or slightly decreased at presentation. Neutrophils often decline with age, and in adult survivors neutropenia occasionally is severe enough to predispose to fatal infection.38
Erythrocyte adenosine deaminase level is elevated in approximately 75 percent of patients but also may be increased in other aregenerative anemias of childhood.39 Serum erythropoietin level, serum iron level, and total iron-binding capacity are high. Ferritin level increases as patients receive multiple transfusions and develop iron overload if they are not treated with chelation therapy.
The characteristic triad consists of the clinical diagnostic features of anemia, reticulocytopenia, and a paucity or absence of erythroid precursors in the marrow. These findings may be supplemented by increased activity of red cell adenosine deaminase and ribosomal gene mutation analysis. FA can be excluded by cytogenetic analyses under clastogenic stress and determination of FA gene mutations (Chap. 35). Transient erythroblastopenia of childhood, which unusually occurs in the first year of life, is established by spontaneous recovery. When presentation occurs at older ages, the distinction between inherited and acquired aplastic anemia can be difficult40: family history, physical anomalies, and characteristic cytogenetic enzymatic, or genetic findings implicate and indicate an inherited disorder.
Untreated inherited pure red cell aplasia is fatal; death results from severe anemia and congestive heart failure. Transfusions, glucocorticoids, and allogeneic stem cell transplantation are of proven efficacy.9,41 Predictors of glucocorticoid administration in glucocorticoid responsive patients include older age at presentation, a family history, and a normal platelet count. Younger age at presentation and premature birth correlate with continued red cell transfusion dependence.42 Supportive care consists of red cell transfusions. To avoid transfusional hemosiderosis, chelation with desferrioxamine should be initiated early (Chap. 43). Injury to visceral organs from iron overload has been a major cause of death in the past.
Red cell transfusions should be leukocyte depleted to avoid alloimmunization (see Chap. 138). Erythrocytes are administered with the goal of eliminating symptoms and permitting normal growth and sexual development, usually achieved by maintaining hemoglobin levels between 7 and 9 g/dL (70 to 90 g/L).
Glucocorticoids are effective in many patients.43 Although the mechanism of action of glucocorticoids in this disease is not understood, their toxicities are substantial, and a response is not predictable. Once the diagnosis is established, prednisone is administered orally at 2 mg/kg daily in three or four divided doses.35,44,45 A reticulocyte response is seen in the majority of patients 1 to 4 weeks later, followed by a rise in hemoglobin level. Once the hemoglobin level reaches 9 to 10 g/dL (90 to 100 g/L), very slow reduction of the glucocorticoid dose is undertaken by decreasing the number of daily doses. When a single daily dose is achieved, an alternate-day schedule is adopted. In general, severe anemia can be avoided with continued glucocorticoid administration. The maintenance dose may be low (1 to 2 mg/day). Some patients may tolerate complete withdrawal of prednisone, but relapse is frequent and most responders become glucocorticoid dependent. A variety of patterns of response have been described, ranging from prompt recovery and apparent cure to refractoriness after a long period of responsiveness.35 Conversely, a second trial of glucocorticoids years after an apparent therapeutic failure may be successful. In a series of 76 patients followed for many years, 59 were treated with prednisone; 31 initially responded, and two of the 25 who initially failed later responded.44 Glucocorticoid responsiveness is strongly associated with better survival, and patients who require low doses of prednisone, or those few who spontaneously remit, may have normal life expectancies. Long-term use of high-dose prednisone results in significant toxicity, including some combination of growth retardation, cushingoid facies, buffalo hump, osteoporosis, aseptic necrosis of the hip and fractures, diabetes, hypertension, and cataracts. Red cell transfusions with iron chelation may be preferable to such an outcome.
Allogeneic stem cell marrow transplantation, when successful, is curative (Chap. 23), but the procedure has not been widely applied to children responding to medical measures. The median life expectancy of patients requiring transfusions and iron chelation is 30 to 40 years. A less-favorable outcome is related to poor compliance and cardiac and hepatic disease from iron overload.46 Because of the morbidity and mortality associated with allogeneic stem cell transplant, most patients have been transplanted late in their disease course, after large numbers of transfusions, accumulation of heavy iron loads, and alloimmunization. Despite the poor predictive factors, 15 of 19 patients of the first published series of cases survived 5 months to many years posttransplant.37 Comparable survival rates have been reported from European47 and Japanese registries.48 Stem cell transplantation from unrelated stem cell donors or use of cord blood stem cells47,49 has been less successful. Recurrent red cell aplasia despite full engraftment was reported in one child after transplantation.50
Other therapies have not gained wide acceptance despite promising pilot studies, including interleukin-3 (IL-3),51 high-dose methylprednisolone,52 cyclosporine and other immunosuppressive agents,53,54 and prolactin induction by metoclopropamide.55 Leucine, which is effective in animal models,56 is in clinical trials.
With better survival, the risk of late development of leukemia has become apparent.14,43 Four of 76 patients followed at Children’s Hospital in Boston died of acute myelogenous leukemia, with a calculated relative risk of greater than 200 times expected.44
Gene transfer in vitro has functionally corrected cells defective in RSP19,57 and in animal models corrected cells show improved erythropoiesis and a survival advantage in vivo58 offering the possibility of gene therapy.
TRANSIENT APLASTIC CRISIS AND TRANSIENT ERYTHROBLASTOPENIA OF CHILDHOOD
Temporary failure of erythropoiesis is clinically identical to pure red cell aplasia except for spontaneous resolution of symptoms and of the laboratory findings of normocytic and normochromic anemia and marrow erythroid hypoplasia, usually over the course of a few weeks. Erythrocyte production is halted: (1) by acute primate erythroparvovirus 1 (B19 parvovirus) infection, typically in the context of underlying hemolytic disease (called transient aplastic crisis); (2) in normal children, usually after an infection by another [unknown] childhood virus (transient erythroblastopenia of childhood); or (3) as a transient reaction to a drug.
An anemic crisis was described in the 1940s first by Lyngar59 and then by Owren,60 Gasser,3 and Dameshek and Bloom61 in kindreds with hereditary spherocytosis. Several children within a family suffered anemic crises and exhibited low, rather than the usually high, reticulocyte numbers. Transient aplastic crisis also was noted as a complication of sickle cell disease.62,63 Marrow examination showed decrease or absence of erythroid precursor cells, and often giant erythroblasts.60,61 An infectious etiology was suspected from the history of a preceding febrile illness in families and its simultaneous occurrence in siblings. After the serendipitous discovery of B19 parvovirus in a normal blood donor, Pattison and colleagues screened large numbers of stored sera for evidence of recent infection. Immunoglobulin (Ig) M antibody or viral antigen was found in the blood of Jamaican children in London, all of whom had transient aplastic crisis of sickle cell disease.64 B19 parvovirus later was established as the agent also responsible for fifth disease.65 In the large cohort of sickle cell patients in Jamaica reported by Serjeant and colleagues,66,67 virtually all episodes of transient aplastic crisis could be linked to B19 parvovirus. In retrospect, red cell aplasias blamed on kwashiorkor, vitamin deficiency, bacterial infections, and chemical exposures likely represented parvovirus infection.
Gasser3 described erythroblastopenia in normal children who ultimately recovered60; the disease was recognized as an entity by Wranne68 in the 1970s. Transient erythroblastopenia of childhood has an unclear etiology but may represent a postviral immune-mediated syndrome. The syndrome is rare and may be declining in incidence.69
B19 parvovirus, a small DNA virus, commonly infects humans. Most of the adult population has IgG antibodies specific to B19.65 The virus is tropic for erythroid progenitor cells,70 mainly because of their P antigen or globoside, the receptor for entry of B19 into the cell71,72 (Fig. 36–1). Infection lyses the target cell and abrogates erythropoiesis in vitro and in vivo. Reticulocytopenia probably accompanies B19 parvovirus infection in all infected persons.73 Anemia only manifests if red cell survival is decreased. Infection ordinarily is terminated by production of neutralizing antibodies to the virus (when such antibodies are absent, persistence of the virus produces chronic pure red cell aplasia). B19 parvovirus may appear in epidemics of fifth disease in the normal population and of transient aplastic crisis, for example, in hematology clinics specializing in sickle cell disease.74,75 In fifth disease, IgM antibody is present in the blood, and virus levels are either low or are not detectable. Symptoms and signs of a typical “slapped cheek” cutaneous eruption and arthralgia or arthritis are secondary to antibody–virus immune complex deposition.
Figure 36–1.
A and B. Giant early erythroblast precursors in the marrow aspirate of a patient with chronic pure red cell aplasia secondary to persistent B19 parvovirus infection. Note the nuclear inclusions (darker nuclear shading) representing parvovirus infection. C. Marrow biopsy section. The arrows point to binucleate erythroid precursor cell with nuclear inclusions representing parvovirus infection. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
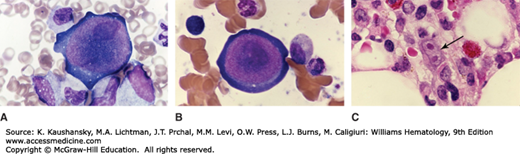
In contrast, in transient aplastic crisis, high concentrations of virus are present in the circulation, and patients do not develop fifth disease. In children with sickle cell disease, the incidence of B19 parvovirus infection was estimated at approximately 11 percent, and 75 percent of patients were infected by age 20 years.76 In this setting, parvovirus infection was associated with transient aplastic crisis, a higher frequency of fever, pain, acute chest syndrome, and acute splenic sequestration syndrome.76 As in normal individuals, parvovirus infection can be asymptomatic in sickle cell disease.77
The origins of transient erythroblastopenia of childhood are poorly understood. An apparent viral prodrome is typical,78 and temporal and seasonal clustering of cases may occur.79,80,81 With rare exception,82 B19 parvovirus is not the etiology,83,84 and no other virus has been consistently implicated.78 Erythroid colony numbers (Chap. 32) usually are low.85 An immune pathophysiology has been inferred from in vitro experiments in which IgG from sera of patients inhibited erythropoiesis86 in the majority of cases.87 Cell-mediated mechanisms also may play a causal role. In one report, T-cell depletion led to a dramatic increase in CFU-E formation.88 A possible relationship between transient erythroblastopenia of childhood and inherited red cell aplasia has been suggested by the clustering of polymorphic alleles in familial transient erythroblastopenia.89
The same drugs implicated in chronic pure red cell aplasia apply to transient erythropoietic failure.90 Laboratory investigations of red cell aplasia secondary to diphenylhydantoin91 and rifampicin92 are consistent with a hapten mechanism, in which serum antibody affects erythroid progenitor cells only in the presence of drug.
Transient aplastic crisis typically occurs in younger patients who are chronically anemic as a result of hereditary spherocytosis, sickle cell disease, or another hemolytic anemia. The decrease in erythropoiesis results in more evident pallor, fatigue on exertion or at rest, lassitude, and dyspnea on exertion. Gastrointestinal complaints or headache may be associated.93 Parvovirus infection can unmask previously undiagnosed underlying hemolytic anemia. Physical examination may reveal signs of anemia, such as pallor, tachycardia, and a flow murmur. No rash or joint swelling is seen. Elevated serum bilirubin or overt icterus may be a clue to underlying hemolysis.
Transient erythroblastopenia of childhood presents as an acute anemia in a previously well child. The syndrome has an estimated incidence rate of 4 to 5 cases per 1 million children.94,95,96 Transient erythroblastopenia is a frequent diagnosis in children with severe anemia,95,97 and is the most common cause of acquired red cell aplasia in pediatric patients.94,98 Most patients are 1 to 3 years old,97,98 but transient erythroblastopenia of childhood can occur in the first year of life and through adolescence. Rare complications include seizures and transient neurologic abnormalities.99,100,101
In both syndromes, anemia is the hallmark, and hemoglobin levels may be markedly depressed. Reticulocytes usually are absent from the blood, and erythroid precursor cells are not present or markedly decreased in the marrow. Red cell indices are normal. White blood cell and platelet counts are normal or elevated. Occasionally, neutropenia and thrombocytopenia of mild or moderate degree are present (especially if splenic function is intact, as in hereditary spherocytosis and in transient erythroblastopenia of childhood).97 If the episode is brief and diagnosed during marrow recovery, patients may present with reticulocytosis, and nucleated red blood cells may be seen on the blood film.
The reticulocyte count readily distinguishes the cause of increasing anemia in a patient with hemolytic disease as transient aplastic crisis. The most important differential diagnosis for transient erythroblastopenia of childhood is inherited pure red cell aplasia. For the former, the age at presentation is older, the patient usually has no family history (but transient erythroblastopenia of childhood may be familial and can occur simultaneously in siblings),102 physical anomalies are absent, and the syndrome resolves spontaneously.
In transient erythroblastopenia of childhood (in contrast to inherited red cell aplasia), erythrocyte adenosine deaminase levels are normal, and red cells do not show “stress” patterns of fetal hemoglobin and i antigen (red cell antigen expressed primarily on feral erythrocytes) expression. The patient’s medical history, the red cell indices, and appropriate serum assays should allow prompt exclusion of more common causes of anemia in children, such as iron deficiency or other nutritional deficiencies. When transient erythroblastopenia is associated with neutropenia, acute lymphoblastic leukemia and aplastic anemia may be suspected: marrow examination clarifies the diagnosis.103 A record of current medications, more important in adults, may provide the basis for a tentative diagnosis of drug-induced rather than idiopathic disease.
Transient aplastic crisis resolves as neutralizing antibodies to B19 parvovirus are made, usually within 1 to 2 weeks of infection. Ensuing reticulocytosis may be brisk, and the hemoglobin may transiently rise to higher-than-normal values. White cell and platelet numbers may “rebound,” and some bone pain from marrow expansion may be present. Severe anemia may require transfusion of red blood cells (Chap. 138). No established role for administration of immunoglobulin exists.
Transient erythroblastopenia of childhood typically terminates after a few weeks, but anemia may persist occasionally for months.98 Transfusions may be required during that interval. Overtreatment of a self-limited illness and misdiagnosis of a more serious disease are to be avoided.
For drug-associated transient failure of erythropoiesis, the suspected offending drug is discontinued and the diagnosis established from subsequent clinical improvement.
ACQUIRED PURE RED CELL APLASIA
Related posts:
 Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Hematopoietic Stem Cells, Progenitors, and Cytokines
Examination of the Marrow
Therapeutic Apheresis: Indications, Efficacy, and Complications
Hemolytic Anemia Resulting from Infections with Microorganisms
Structure and Composition of the Erythrocyte
Disorders of Hemoglobin Structure: Sickle Cell Anemia and Related Abnormalities
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


