INTRODUCTION
SUMMARY
Increased blood red cell mass can be termed either polycythemia or erythrocytosis; no clear consensus for either term has been achieved. Primary polycythemias are caused by acquired or inherited mutations causing functional changes within hematopoietic stem cells or erythroid progenitors leading to an accumulation of red cells. The most common primary polycythemia, polycythemia rubra vera, which is a clonal disorder, is discussed in Chap. 84; other primary polycythemias, such as those inherited from mutations in the erythropoietin receptor or congenital disorders of hypoxia sensing, are discussed herein. In contrast, secondary polycythemias are a result of either an appropriate or inappropriate increase in the red cell mass, most often as a result of augmented levels of erythropoietin; these polycythemias can also be either acquired or hereditary. Although the clinical presentations of primary and secondary polycythemias may be similar, distinguishing amongst them is important for accurate diagnoses and proper management.
For example, those secondary polycythemic states that represent an appropriate physiologic compensation to tissue hypoxia, should not be treated by phlebotomies. An occasional patient may experience hyperviscosity symptoms and may benefit from isovolemic reduction of hematocrit. Inappropriate polycythemias are caused by erythropoietin-secreting tumors, self-administration of erythropoiesis-stimulating agents, including androgens, inherited disorders of hypoxia sensing, or, rarely, some endocrine disorders (described in Chap. 38). Correction of hypoxia, discontinuation of erythropoiesis-stimulating agents or resection of erythropoietin-secreting tumors will typically correct the associated polycythemia.
Acronyms and Abbreviations:
BFU-E, burst-forming unit–erythroid; 2,3-BPG, 2,3-bisphosphoglycerate; COPD, chronic obstructive pulmonary disease; EGLN1, a gene encoding for PHD2; EPAS1, a gene encoding for HIF-2α; EPOR, erythropoietin receptor (protein); HCP, hematopoietic cell phosphatase; HIF, hypoxia-inducible factor; HUMARA, human androgen-receptor gene; JAK, Janus-type tyrosine kinase; OSA, obstructive sleep apnea; PAI-1, plasminogen activator inhibitor; PFCP, primary familial and congenital polycythemia; PHD2, proline hydroxylase 2; STAT, signal transducer and activator of transcription; VEGF, vascular endothelial growth factor; VHL, von Hippel-Lindau syndrome.
DEFINITION AND HISTORY
The term polycythemia, denoting an increased amount of blood cells, has traditionally been applied to those conditions in which the mass of erythrocytes is increased. Erythrocytosis is an alternative term that has also been applied to circumstances in which the increased red cell mass is the singular finding, distinguishing it from polycythemia vera in which, classically, there is an increase in red cells, granulocytes, and platelets. Although this usage has much to recommend it, no consensus about terminology has been reached and the term polycythemia is used interchangeably with erythrocytosis by many physicians. In some instances, time-honored terms such as post–renal transplant erythrocytosis or Chuvash polycythemia will be used. A classification of the polycythemias is presented in Chap. 34 in Table 34–2.
Polycythemia vera (Chap. 84) and primary familial and congenital polycythemia (PFCP) are primary polycythemic disorders, which have erythroid progenitors that are hypersensitive to erythropoietin.1,2,3 These are caused by somatic (polycythemia vera) or germline (PFCP) mutations wherein erythroid progenitors are intrinsically hyperproliferative by mechanisms other than the presence of increased levels of erythropoietin and have, as shown by in vitro assays, an augmented response to erythropoietin. Some congenital polycythemias, as best described in Chuvash polycythemia, have erythroid progenitors that are hypersensitive to erythropoietin, but also may have normal or even increased erythropoietin levels despite the increased red cell volume.4,5 Thus, some of these rare inherited polycythemias share features of both primary and secondary polycythemias.6
The term secondary polycythemia, more appropriately secondary erythrocytosis, which refers to those conditions in which only erythrocytes are increased in number and volume, describes a group of disorders characterized by an increased red cell mass brought about by enhanced stimulation of red cell production by circulating physiologic mediators, most commonly erythropoietin. Secondary polycythemia may be subdivided into appropriate polycythemia, that is, responding normally to tissue hypoxia such as in pulmonary disease, Eisenmenger complex, high-altitude polycythemia and hemoglobins with increased affinity for oxygen (Chaps. 49 and 50), and inappropriate polycythemia in which erythropoiesis is being stimulated by the aberrant production of erythropoietin, as in erythropoietin-secreting tumors, by high levels of insulin growth factor 1, by cobalt toxicity, by self-administration of erythropoietin, androgens, or adrenocorticotropic hormone, or by other stimulators of erythropoiesis (as in post–renal transplant erythrocytosis).7
In his important monograph on barometric pressure published in 1878, Paul Bert showed that physiologic impairment observed at high altitude was caused by a reduction in the oxygen content of the air.8 A few years earlier, his friend and mentor, Dennis Jourdanet, had observed an increase in the number of red corpuscles in blood of the highlanders in Mexico,9 and Bert recognized that such an increase would tend to ameliorate the effect of atmospheric hypoxia. However, neither Bert nor Jourdanet suspected a cause-and-effect relationship. It was not until 1890, when Viault10 observed a prompt increase in the number of his own red corpuscles after having traveled from Lima, Peru, at sea level, to Morococha, at 4570 m (15,000 ft) above sea level, that altitude erythrocytosis was accepted as a compensatory adaptation to hypoxia.11 At about the same time, it was observed that many patients with cyanosis were also polycythemic. Both cardiacos negros,12 with severe pulmonary failure and arterial oxygen desaturation, and children with morbus caeruleus, or right-to-left shunt through a congenital cardiac malformation, were found to have increased red cell counts.13 Mechanical or neurogenic hypoventilation as a cause of cyanosis and polycythemia was first popularized in 1956 with the classic description of the pickwickian syndrome by Burwell and colleagues.14 Polycythemia associated with carboxyhemoglobinemia resulting in hypoxemia as a result of smoking and with tissue hypoxia because of inherited abnormal hemoglobins with high-affinity oxygen binding to hemoglobin was recognized subsequently (Chaps. 49 and 50).15 Erythrocytosis associated with abnormal hemoglobins with an increased affinity for oxygen also represents an appropriate response to hypoxia first noted by Charache and colleagues15 in 1966 when they described hemoglobin Chesapeake.
Relative erythrocytosis is the term used to depict enhanced red cell count or blood hemoglobin values resulting from reduced plasma volume, not increased red cell mass. The disorder is, therefore, not a true polycythemia and is designated apparent, spurious, or relative polycythemia. The cause of the reduced plasma volume and hence relative erythrocytosis is often known, that is, diuretic use, dehydration from excessive sweating, etc. However, there are some patients with mild erythrocytosis in which neither the cause nor the clinical significance is clear. In 1905, Gaisbock reported that a number of hypertensive patients had plethora and an elevated red cell count but no splenomegaly, a condition he termed polycythemia hypertonica, sometimes called Gaisbock syndrome.16 In 1952, direct measurement of blood volume in patients with polycythemia led Lawrence and Berlin to identify a subgroup of patients with a normal red cell volume but reduced plasma volume. Although some members of this group were hypertensive, the authors were more impressed by their tense and anxious behavior and coined the term stress polycythemia.17
EPIDEMIOLOGY
This autosomal dominant disorder (designated PFCP) is uncommon but more prevalent than is generally appreciated, as many affected subjects are initially misdiagnosed as having polycythemia vera. Its prevalence is similar to congenital polycythemias because of high oxygen-affinity hemoglobin mutants, and far more common than 2,3-bisphosphoglycerate (2,3-BPG) deficiency.18
In one study of 2524 patients with severe chronic obstructive pulmonary disease (COPD), 8.4 percent had a hematocrit higher than 55 percent. In this study, hematocrit was an independent predictor of longer survival, decreased hospital admission rate, and decreased cumulative duration of hospitalization.19 In another, smaller study of 309 subjects with COPD and chronic respiratory failure, 67 percent had normal hemoglobin levels, 20 percent had anemia, and 18 percent had polycythemia.20
Although the evidence is largely anecdotal,21 secondary polycythemia is a widely recognized complication of longstanding obstructive sleep apnea (OSA), being found in 5 to 10 percent of those with nocturnal apnea and hypopnea.22 The published studies remain controversial; in a study of 263 patients (189 men and 74 women), patients with severe sleep apnea had significantly higher hematocrit values than did patients with mild to moderate sleep apnea or nonapneic controls (p <0.01).23 In contrast, in other studies, there were no significant differences in hemoglobin levels or hematocrit between subjects with and without OSA.24,25 The total number of hours of hypoxia per day may dictate whether the stimulus to erythropoietin production is sufficient to cause erythrocytosis.
Smoking clearly increases hematocrit in COPD compared to controls of comparable pulmonary function. In a study of 2524 patients with severe COPD, 10.2 percent of patients reported as current smokers had significantly higher hematocrit values than did ex-smokers or nonsmokers of comparable pulmonary impairment of both genders, with p <0.02.20
Even young male smokers without pulmonary impairment have higher hematocrits. In one study, 1169 subjects (age range: 18.6 to 22.8 years; mean: 19.4 years) were recruited and 25 percent were smokers. Predictably, carboxyhemoglobin was much higher in smokers than in nonsmokers (r = 0.958, p <0.001) and both hemoglobin and hematocrits were also markedly higher in smokers (hemoglobin (p = 0.001), hematocrit (p = 0.004).26
These disorders are reviewed in detail in Chaps. 49 and 50. High oxygen-affinity hemoglobins deliver less oxygen to tissues, which is appropriately compensated by increased erythropoiesis and a higher steady-state hemoglobin concentration. While considered rare, high oxygen affinity has been found, according to one report, in approximately 20 percent of 70 unrelated subjects with otherwise idiopathic polycythemia.27
Eisenmenger syndrome, characterized by elevated pulmonary vascular resistance and right-to-left shunting of blood, is usually accompanied by polycythemia.28 Most patients with the syndrome survive for 20 to 30 years.
Erythrocytosis has been reported in Cushing syndrome,29 primary aldosteronism,30 and Bartter syndrome (Chap. 38).31
In a prospective trial of testosterone use in older men, erythrocytosis (defined as a hematocrit greater than 50 or 52 percent) was three times more likely to occur in the testosterone-treated group compared to placebo.32 (Refer to Chap. 38 for more details.)
The prevalence of various types of secondary polycythemia is a function of underlying causes, such as geographical location of the patient or presence of a causative neoplasm. Approximately 1 to 3 percent of all patients with pheochromocytoma or paraganglioma have erythrocytosis.33 Rare patients with congenital erythrocytosis will develop pheochromocytoma or paraganglioma.34 Uterine leiomyomas in premenopausal women are very common, estimated at 20 to 40 percent, and the occurrence of erythrocytosis ranges from 0.02 to 0.5 percent of cases.35 Isolated instances of polycythemia have been attributed to myxoma of the atrium,36 hamartoma of the liver,37 and focal hyperplasia of the liver.38 Erythrocytosis and inappropriate secretion of erythropoietin may be found in approximately 15 percent of patients with cerebellar hemangioma.39,40
Athletes have attempted for decades to manipulate their blood to gain a competitive advantage either by blood transfusions or erythropoietin. This evolving, but continuous, problem has been the subject of a review.41
Elevation of red cell mass as a response to high altitude hypoxia represents an appropriate adjustment to reduced blood oxygen content and delivery. The exponentially decreasing atmospheric oxygen pressure with altitude stimulates the body to accommodate by an increase in respiratory rate and volume. Such adaptation is possible only in the short-term, because the body may not always be able to adequately enhance respiration. Polycythemia is considered to be a universal, uniform adaptation response to hypoxia that arises in normal individuals, but when it is exaggerated, in some cases it results in chronic mountain sickness with associated symptoms of fatigue, headache, and pulmonary hypertension.42
Altitude above sea level should be used as an independent variable for the definition of polycythemia43 and Centers for Disease Control data lists the appropriate adjustment values.44
This syndrome, defined as a persistent elevation of hematocrit at greater than 51 percent, is a relatively common condition found in approximately 5 to 10 percent of renal allograft recipients.45,46 Post–renal transplant erythrocytosis usually develops within 8 to 24 months after transplantation, despite persistently good function of the allograft, and resolves spontaneously within 2 years in approximately 25 percent of patients.47 Factors that increase the likelihood of its development are lack of erythropoietin therapy prior to transplantation, a history of smoking, diabetes mellitus, renal artery stenosis, low serum ferritin levels, and normal or higher pretransplant erythropoietin levels. Post–renal transplant erythrocytosis is also more frequent in patients who are not undergoing graft rejection.
A Russian hematologist, Lydia A. Polyakova, described polycythemia in the Chuvash population (an ethnic isolate in the mid-Volga River region of Russia of Turkish descent) in the early 1960s,48 and by 1974, 103 cases from 81 families had been described.48 Since then, more cases have come to light; hundreds of children and adults suffer from this condition, indicating that Chuvash polycythemia is the only known endemic congenital polycythemia in the world.49 Outside of Chuvashia, Chuvash polycythemia has also been found sporadically in diverse ethnic and racial groups,50,51 and recently a high prevalence of this disorder has also been reported on the Italian island of Ischia.52
ETIOLOGY AND PATHOGENESIS
In contrast to polycythemia vera, PFCP is caused by germline rather than acquired somatic mutations. It is congenital and manifests autosomal dominant inheritance3 and, not infrequently, sporadic occurrence from de novo germline mutations. Like polycythemia vera (Chap. 84), it is primary in that the defect changes intrinsic responses of erythroid progenitors, and erythropoietin levels are low.
To date, 12 mutations of the erythropoietin receptor (EPOR) associated with PFCP have been described (Table 57–1). Nine of the 12 result in truncation of the EPOR cytoplasmic carboxyl terminal, and are the only mutations convincingly linked with PFCP. Such truncations lead to a loss in the negative regulatory domain of the EPOR (Chaps. 32 and 34). Three missense EPOR mutations have also been described, but these have not been linked to PFCP or any other disease phenotype (Table 57–1).
| Type of Mutation | Mutation | Structural Defect | Association with PFCP | Ref. |
|---|---|---|---|---|
| Deletion (7bp) | Del5985–5991 | Frameshift > ter truncation | Yes | 163,192 |
| Duplication (8 bp) | 5968–5975 | Frameshift > ter truncation | Yes | 222 |
| Nonsense | G6002 | Trp439 > ter truncation | Yes | 223 |
| Nonsense | 5986 C>T | Gln435 > ter truncation | Yes | 224 |
| Nonsense | 5964C>G | Tyr426 > ter truncation | Yes | 162 |
| Nonsense | 5881C>T | Glu399 > ter truncation | Yes | 225 |
| Nonsense | 5959G>T | Glu425 > ter truncation | Yes | 226 |
| Insertion (G) | 5974insG | Frameshift > ter truncation | Yes | 227 |
| Insertion (T) | 5967insT | Frameshift > ter truncation | Yes | 228 |
| Substitution | 6148C>T | Pro 488 > Ser | No | 192,229 |
| Substitution | 6146A>G | Asn487 > Ser | No | 230 |
| Substitution | 2706 A>T | Unknown | No | 226 |
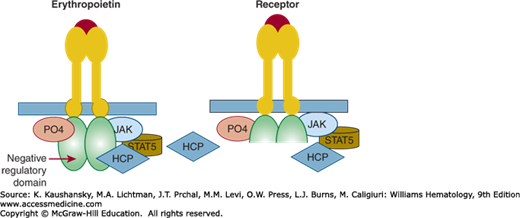
Erythropoietin-mediated activation of erythropoiesis involves several steps (also see Chap. 32). First, erythropoietin activates its receptor by inducing conformational changes of its dimers (Chap. 17). These changes lead to initiation of an erythroid-specific cascade of events. The first signal is initiated by conformation change-induced activation of Janus-type tyrosine kinase 2 (JAK2) and its phosphorylation and activation of a transcription factor, signal transducer and activator of transcription 5 (STAT5), which regulates erythroid-specific genes. This “on” signal is negated by dephosphorylation of EPOR by hematopoietic cell phosphatase (HCP), also known as SHP1, that is, the “off” signal. EPOR truncations lead to a loss of the negative regulatory domain of the EPOR, a binding site for HCP, explaining the gain-of-function properties of these EPOR mutations (Fig. 57–1).
Figure 57–1.
Left panel: Erythropoietin binding to a normal erythropoietin receptor (EPOR) results in interaction of a protein kinase (JAK) with the receptor. The interaction leads to phosphorylation of the receptor and initiates a cascade of signaling that ultimately results in erythroid progenitor proliferation and differentiation. This process is self-regulatory. Activated signal transduction molecules, hematopoietic cell phosphatase (HCP) binds to the C-terminal of the EPOR, which is a negative regulatory domain. This interaction dephosphorylates the receptor and turns off the signaling resulting in cessation of erythroid progenitor proliferation. Right panel: Patients with mutated gain-of-function EPOR gene lack the C-terminal portion of the receptor that contains the negative regulatory domain. Erythropoietin binds and the signal transduction pathway is activated by change of configuration of erythropoietin receptor dimer, but because there is there is no structure for HCP to bind on the activated EPOR dimer, the receptor is left in the activated position resulting in unbridled erythroid proliferation and an elevated red cell mass. PO4, phosphate; STAT, signal transducer and activator of transcription.
The morbidity of primary polycythemias, such as polycythemia vera (Chap. 84), is largely the result of increased activated neutrophils and, perhaps, attendant pathologic platelet–endothelial interactions, whereas in secondary polycythemias it is presumably related to an increase in blood viscosity and, in part, to the resulting increased cardiac work.53 In most instances, the etiology of morbidity or mortality, such as associated with congenital disorders of hypoxia sensing, is largely unknown.54 The effect of blood viscosity on oxygen delivery is often oversimplified and the emphasis on the hematocrit alone may lead to ill-advised therapeutic interventions. In the normovolemic state, viscosity increases in a log-linear fashion as hematocrit increases, and the effect is particularly pronounced when the hematocrit rises above 50 percent. The prediction is that oxygen delivery decreases as hematocrit rises significantly above 50, as the greatly increased viscosity reduces blood flow, overshadowing the increased oxygen-carrying capacity of blood with a higher concentration of hemoglobin. However, polycythemia is not a normovolemic state, but is accompanied by an increase in blood volume, which, in turn, enlarges the vascular bed and decreases peripheral resistance (Chap. 34). Thus, hypervolemia can increase oxygen transport, and the optimum for oxygen transport occurs at higher hematocrit values than in normovolemic states. Consequently, despite the attendant increase in viscosity, an increase in hematocrit may generally be of benefit in appropriate secondary polycythemias. However, at some point, the high viscosity causes an increase in the work of the heart and a reduction in blood flow to most tissues and may be responsible for cerebral and cardiovascular impairment.
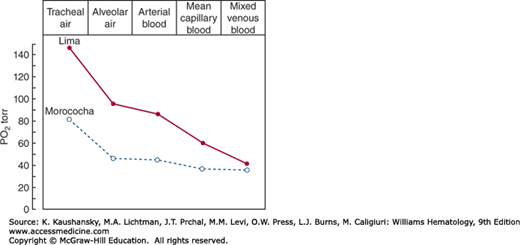
Adaptive adjustments of humans living at high altitude involve a series of steps that reduce the steepness of the oxygen gradient between the atmosphere and mitochondria (Fig. 57–2).55 The initial oxygen gradient between atmospheric and alveolar air can be reduced by an increase in respiratory rate and volume. Because dead space and water vapor pressure are constant and acclimatized individuals do not ventilate excessively, the normal sea level gradient of about 60 torr is only reduced to approximately 40 torr at 4540 m (14,900 ft) above sea level.55 Further reduction can be achieved, and at the top of Mount Everest, extreme hyperventilation reduces the gradient to less than 10 torr. A shift in the oxygen dissociation curve to the right, which represents decreased affinity of hemoglobin for oxygen, may be of benefit for short-term high-altitude acclimatization,56 but its usefulness for chronic acclimatization has probably been exaggerated.57 In the unacclimatized subject exposed acutely to high altitude, hyperventilation alkalosis leads initially to a shift of the oxygen dissociation curve to the left, representing an increased affinity of hemoglobin for oxygen, further worsening already present tissue hypoxia. The alkalosis and the hypoxia will, in turn, promote red cell synthesis of 2,3-BPG and cause the oxygen dissociation curve to shift back to a normal or even right-shifted position (Chap. 49). In chronic acclimatization, blood pH is slightly increased, and when this is taken into account, the dissociation curve is shifted approximately to normal.58 It is unlikely that a shift to the right would be to the advantage of high-altitude dwellers, except as a partial compensation for respiratory alkalosis.59 In addition, a right-shifted curve also has a decrease in oxygen loading in the alveolar capillary, minimizing any net gain in off-loading. There is a relationship between higher altitude and hemoglobin concentration response, best studied among Andean highlanders and Europeans in the United States; hemoglobin concentration is almost 10 percent higher in those Andean highlanders living at 5500 m than in those living at 4355 m. Furthermore, native Andean high-altitude dwellers have a gradual increase in their hemoglobin levels with age60 and body weight.61 Although it has been postulated that high hemoglobin–oxygen affinity in the setting of extremely low ambient oxygen may be one such adaptive change,62 increased hemoglobin–oxygen affinity or increased fetal hemoglobin are not adaptive phenotypes of Tibetan or Andean highlanders.63
In a subset of Andean high-altitude native dwellers, namely Quechua and Aymara Indians, polycythemia becomes excessive and often results in chronic mountain sickness with its associated constitutional symptoms and pulmonary hypertension.42,60 This excessive erythrocytosis, called Monge disease or chronic mountain sickness,42,64 is also described in Han Chinese living in Tibet65 and occurs in whites living at high altitudes.66
The polycythemia encountered in high-altitude dwellers is often considered to be a universal, uniform adaptation process to hypoxia that would arise in all normal individuals. In reality, there is marked variability in erythropoietin level and subsequent polycythemic response to chronic hypoxia,60,67 suggesting that some of these factors may be genetically determined; the same degree of hypoxia induces substantial differences in erythropoietin production in response to high altitude.62,68,69 Three distinct adaptations to high altitude appear to have evolved. Andean highlanders have higher oxygen saturation than Tibetans at the same altitudes.62 Tibetan mean resting ventilation and hypoxic ventilatory response are higher than Andean Aymaras, whereas the mean Tibetan hemoglobin concentration is below the Andean mean. High levels of nitric oxide (NO) in the exhaled breath of Tibetans may represent increased physiologic NO. This effect may improve oxygen delivery by inducing vasodilation and increasing blood flow to tissues, thus making the compensatory increase in red cell volume unnecessary.70 Another distinct successful pattern of human adaptation in high-altitude dwellers that contrasts with both the Andean “classic” (arterial hypoxemia with polycythemia) and Tibetan (arterial hypoxemia with normal venous hemoglobin concentration) patterns evolved in Ethiopia. While Ethiopian high-altitude dwellers have hemoglobin concentrations that fall in the normal range (15.9 and 15.0 g/dL for males and females, respectively), they have a surprisingly, as-yet-unexplained high (average of 95.3 percent) oxygen saturation of hemoglobin despite their hypoxic environment (reviewed in Ref. 62). Their cerebral circulation is increased but is insensitive to hypoxia, unlike Peruvian high-altitude dwellers.71 Thus, Ethiopian highlanders maintain venous hemoglobin concentrations and arterial oxygen saturation within the ranges of sea level populations, despite the decrease in ambient oxygen tension at high altitude.72 Tibetans and Ethiopians have lived as mountain dwellers much longer than the Quechua or Aymara Indians,73 suggesting that extreme elevation of red cell mass is a maladaptation that Tibetans avoided by evolving a more efficient, or less detrimental, compensatory mechanism than that which causes Monge disease.
With rapid advances in genomics (Chap. 11), progress has been made in identification of the molecular basis of high-altitude adaptation; most of these advances have been in our understanding of Tibetan adaptation. Several studies reported evidence for positive natural selection in genomic regions in Tibetans; not surprisingly, most are haplotypes comprising genes that are components of hypoxia sensing that are mediated by hypoxia-inducible factors (HIFs) (described in Chap. 32). Two of these selected regions include genes that have undergone the strongest genetic selection, and are thus likely the most beneficial for Tibetan adaptation. These two regions encompass the EPAS1 gene, encoding the α subunit of HIF-2, and the EGLN1 gene, encoding proline hydroxylase 2 (PHD2). PHD2 is the principal negative regulator of both HIF-1 and HIF-2. Both of these haplotypes were shown to be associated with differences in hemoglobin concentration at high altitude by several independent studies.74,75,76 Intriguingly, the EPAS1 haplotype was previously identified as having the strongest Tibetan positive selection and was found to have an unusual haplotype structure that originated by introgression of DNA from Denisovan or Denisovan-related hominins.77 Denisovans, a sister group to the Neanderthals, branched off from the human lineage perhaps 600,000 years ago,78 and available evidence suggests that Denisovan and Neanderthal hominins contributed to the modern homo sapiens admixture before their extinction, likely by interbreeding,79 and that these hominin species provided genetic variations that helped humans adapt to new environments, such as extreme hypoxia associated with high altitude.77 The first Tibetan adaptation gene mutation identified, which changes the encoded PHD2 protein within the selected haplotype, is a missense variant of the EGLN1 gene, c.12C>G,80 that is in near complete linkage disequilibrium with a previously reported missense variant, EGLN1:c.380G>C.80 Both EGLN1:c.[12C>G; 380G>C] (PHD2D4E,C127S) are in cis; that is, the constituting PHD2D4E,C127S locus. Analysis of Tibetans and related populations suggests that 12C>G started on the 380G>C variant that is not Tibetan specific,80,81 that PHD2D4E,C127S originated from a single individual approximately 8000 years ago,81 and that now greater than 80 percent of Tibetans carry this PHD2 variant.81 Functional assessment of homozygous PHD2D4E,C127S recombinant proteins showed that the variant protein has increased hydroxylase activity under hypoxic conditions. Furthermore, native homozygous PHD2D4E,C127S erythroid progenitors have blunted erythropoietic responses to hypoxia by both erythropoietin-specific and erythropoietin-independent mechanisms.81 Although this is the first identified variant that contributes to the molecular and cellular basis of Tibetan adaptation to high altitude, there are other evolutionarily selected genomic regions, and elucidation of their functional impact is, at the time of this writing, unknown.
Understanding the etiology of polycythemia of high altitude is made more complex by a study of inhabitants of the Peruvian mining community of Cerro de Pasco (altitude 4280 m) with excessive erythrocytosis (mean hematocrit: 76 percent; range: 66 to 91 percent). About half of those individuals with a hematocrit greater than 75 percent had toxic serum cobalt levels,82 suggesting that other erythropoiesis promoting factors such as cobalt83 can augment hypoxia induction of erythropoietin, causing extreme polycythemias (Chap. 32). Most high-altitude dwellers do not have measurable levels or a history of exposure to cobalt or other heavy metals.84
Degrees of arterial hypoxia comparable to those observed in individuals at high altitudes are observed in patients with right-to-left shunting resulting from cardiac or intrapulmonary shunts or to ventilation defects, as in COPD.
Many patients with COPD with severe cyanosis are not polycythemic. This has been attributed to infections and inflammation often present in the lungs, resulting in anemia of chronic inflammation, and to an increase in plasma volume. Why some patients with lung disease and congenital heart disease develop polycythemia, while others do not, is not entirely clear.
Patients with right-to-left shunting (Eisenmenger syndrome) develop a degree of erythrocytosis comparable to that observed with similar degrees of desaturation at high altitudes.85 The hematologic changes associated with this syndrome include hyperviscosity caused by erythrocytosis. Erythrocytosis is present in most patients, but excessive phlebotomy may cause microcytosis and some have attributed this effect to the exacerbation of the symptoms of hyperviscosity.28 In view of recent understanding of physiology of HIF regulation, it may not be the microcytosis, per se, that is detrimental, but the induced iron deficiency that inhibits PHD2 and increases HIF, which then directly causes pulmonary vasoconstriction and enhanced pulmonary vascular pressure (Chaps. 32 and 34).
In the colorfully named pickwickian syndrome,86 polycythemia is characterized by its association with extreme obesity and somnolence. Today, the more widely studied OSA may not always be associated with obesity87 but can, if severe, cause arterial hypoxemia, hypercapnia, somnolence, and secondary polycythemia.88
Heavy smoking will result in the formation of carboxyhemoglobin, which does not transport oxygen (Chap. 50), and causes an increase in oxygen affinity of the remaining normal hemoglobin. Carboxyhemoglobin increases in relationship to the number of cigarettes or cigars smoked each day (Table 57–2). This leads to tissue hypoxia, erythropoietin production, and stimulation of red cell production.89 Smoking may also cause a reduction in plasma volume.90 Either augmentation of red cell mass or shrinkage of plasma volume could easily explain the rise in the hematocrit.
| Subject | Hemoglobin (Hgb) (g/dL) | Carboxyhemoglobin (COHb) (g/dL) | Hgb-COHb (g/dL) | Affinity Correction (g/dL) | Adjusted Hgb (g/dL) |
|---|---|---|---|---|---|
| Healthy male nonsmokers | 16 (14–18) | 0.16 (0.08–0.25) | 15.8 (14–18) | 0 | 16 (14–18) |
| Male smokers with increased Hgb concentration | 20 (17–23) | 2 (1–3) | 1816–21 | 1.5 (0.5–2.0) | 16.5 (15–19) |
Hemoglobins with certain amino acid substitutions manifest an increased affinity for oxygen, producing tissue hypoxia and compensatory erythrocytosis (Chap. 49). Mutations affecting amino acids of the α1β2-globin chain interface affect normal rotation within molecules and impair the rate of deoxygenation. Changes in the carboxyl terminal and penultimate amino acids also impair intramolecular motion and tend to keep molecules in a high-affinity state. Alterations in the amino acids lining the central cavity of hemoglobin destabilize the binding of 2,3-BPG in this cavity and lead to increased oxygen affinity (Chaps. 47 and 49). Finally, some heme pocket mutations interfere with deoxygenation. Most hemoglobins with a mutation involving amino acids in the heme pocket are unstable and are associated with hemolytic anemia and cyanosis. Inheritance of these disorders is autosomal dominant. High-affinity hemoglobins result from mutations in any of three globin genes; those from α-globin gene mutations are congenital and life-long. β-Globin gene mutations are not apparent at birth but manifest after fetal to adult hemoglobin switching at approximately 6 months of life, while γ-globin gene mutations cause transient increase of hemoglobin concentration at birth lasting only about 6 months.
Deficiencies of red cell enzymes in early steps of glycolysis sometimes cause a marked decrease in the levels of 2,3-BPG (Chap. 47). Occasionally, mild polycythemia occurs in patients with methemoglobinemia as a result of cytochrome b5 reductase (methemoglobin reductase) deficiency (Chap. 50).
The same pathophysiology as that seen in high-affinity hemoglobins is also exhibited in mutations of the 2,3-BPG mutase gene, resulting in low 2,3-BPG. Because these mutations are very rare, with only a single family comprehensively studied,91 it is not clear if the mode of inheritance is recessive or dominant.
This condition, as well as other high-affinity hemoglobins, can only be conclusively confirmed by direct measurement of a hemoglobin dissociation curve, conveniently expressed as the partial pressure of oxygen required to saturate 50 percent of hemoglobin (p50O2); when equipment for this is not available, p50 can be estimated from pH, pO2 and hemoglobin oxygen saturation of venous blood.92,93
A number of chemicals have been suspected of causing histotoxic anoxia and secondary polycythemia, but the only chemical with a predictable capacity to cause erythrocytosis is cobalt.83 Cobalt administration increases erythropoietin production by increasing HIFs (see Chap. 32).94
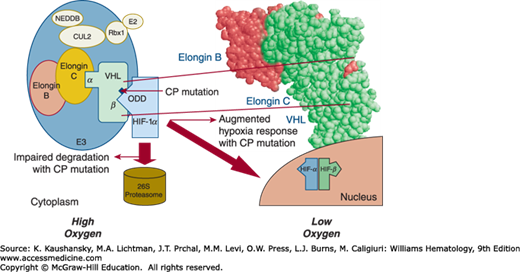
Chuvash polycythemia is the only known endemic congenital polycythemia. The condition is caused by an abnormality in the oxygen-sensing pathway and causes thrombotic and hemorrhagic vascular complications that lead to early mortality; survival beyond age 65 years is uncommon.48,95 Inheritance is autosomal recessive, and affected patients tend to have normal blood gases, normal calculated p50, normal to increased erythropoietin levels, absence of genetic linkage to erythropoietin and EPOR loci, and no evidence of abnormal hemoglobin.95 In a study of five multiplex Chuvash families with Chuvash polycythemia, a homozygous mutation of the von Hippel-Lindau (VHL) gene (598C>T); that is, VHLR200W, was found in all affected individuals. This mutation impairs the interaction of VHL protein (pVHL) with both HIF-1α and HIF-2α, thus reducing the rate of ubiquitin-mediated destruction of HIF-1α and HIF-2α (Chap. 32). As a result, the level of HIF-1 and HIF-2 heterodimers increases and leads to the increased expression of target genes, including the erythropoietin (EPO), vascular endothelial growth factor (VEGF), and plasminogen activator inhibitor genes (PAI-1), among others.4,5 Figure 57–3 depicts the effect of this mutation on hypoxic sensing. The role of circulating erythropoietin in the Chuvash polycythemia phenotype is indisputable. However, there must be other factors associated with the Chuvash polycythemia VHL mutation that contribute to the polycythemic phenotype, as the erythroid progenitors of Chuvash polycythemia patients are hypersensitive in vitro to extrinsic erythropoietin; the mechanism of this observation is not fully explained.4,5 Some, but not all, patients with other VHL mutations have erythropoietin hypersensitive erythroid colonies96,97,98; in these patients there is also increased expression of the RUNX1 and NFE1 genes, which can stimulate erythropoiesis.6
Figure 57–3.
Elongins B, C, and proteins Rbx1, Cul2 E2, and NEDD 8 are interacting proteins that facilitate von Hippel-Lindau (VHL) function. Interaction of mutated VHL protein with HIF-1α. The Chuvash VHL mutation leads to the impaired interaction with HIF-1α, which results in impaired degradation in 26S proteasome and augmented hypoxia sensing. CP, Chuvash polycythemia.
Despite increased expression of HIF-1α, HIF-2α, and VEGF in normoxia, Chuvash polycythemia patients do not display a predisposition to tumor formation. Imaging studies of 33 Chuvash polycythemia patients revealed unsuspected cerebral ischemic lesions in 45 percent, but no tumors characteristic of VHL syndrome.99 There also, is a high prevalence of this disorder on the Italian island of Ischia.52 The Chuvash VHLR200W mutation has also been described in whites in the United States and Europe, and in people of Punjabi/Bangladeshi Asian ancestry.100 Some patients with congenital polycythemia have proved to be compound heterozygotes for the VHLR200W mutation and other VHL mutations. Additionally, two distantly related Croatians with polycythemia are homozygous for VHLH191D, the first example of a homozygous VHL germline mutation other than VHLR200W causing polycythemia.51,54,101,102,103,104,105
A small number of cases of congenital polycythemia that appear to have a mutation of only one VHL allele confound an obvious pathophysiologic explanation. In a Ukrainian family, two children with polycythemia were heterozygotes for VHL 376G>T (D126Y), but the father with the same mutation was not polycythemic.104 An English polycythemic patient was a heterozygote for VHL 598C>T106; but the inheritance of the deletion of a VHL allele, or null VHL allele, in a trans position was not excluded. Subsequently, two polycythemic VHL heterozygous patients were described in whom a null VHL allele was more rigorously excluded101,102; the molecular mechanism of their polycythemic phenotype remains to be elucidated.
To address the question of whether the VHL 598C>T substitution occurred in a single founder or resulted from recurrent mutational events, haplotype analysis of eight highly informative single nucleotide polymorphic markers covering 340 kb spanning the VHL gene was performed on 101 subjects bearing the VHL 598C>T mutation and 447 normal unrelated individuals from Chuvash, Southeast Asian, white, Hispanic, and African American ethnic groups.49 Polymorphism of the VHL locus in normal controls (having a wild VHL 598C allele) and subjects bearing Chuvash polycythemia VHL 598T were in strong linkage disequilibrium. These studies indicated that, in most individuals, the VHL 598C>T mutation arose in a single ancestor between 51,000 and 12,000 years ago. However, this is not the case for a Turkish polycythemic family with a VHL 598C>T mutation wherein the VHL
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


