POLIOMYELITIS, POLIO VACCINES, AND THE POSTPOLIOMYELITIS SYNDROME
JOHN F. MODLIN AND DAVID J. COFFEY
Poliomyelitis is a systemic viral infection caused by any of three human poliovirus serotypes that is characterized by acute meningitis and lytic infection of motor neurons in the brainstem and spinal cord resulting in cranial nerve dysfunction and transient or permanent paresis of one or more extremities. In the United States and other developed countries, poliomyelitis due to naturally occurring polioviruses has been eliminated by routine childhood immunization. Global eradication is a major priority of the World Health Organization (WHO) and other major partners in the Global Poliomyelitis Eradication Initiative.
A BRIEF HISTORY OF POLIOMYELITIS
Poliomyelitis has afflicted humans since antiquity (1). The first acknowledged description in English is a biographical narrative of the acute paralysis suffered by Sir Walter Scott in the second year of his life, circa 1772, and the first medical description is recorded in the second edition of A Treatise on Diseases of Children, published in 1789 by Michael Underwood, a London pediatrician and obstetrician (2,3). Major contributions to understanding the disease were made by the German orthopedist Heine, whose 1840 monograph describes the clinical features of acute poliomyelitis in young children (4), by the French anatomist Charcot who showed that paralysis was accompanied by a loss of motor nerve cells within the anterior horns of the cord (5), and by Karl Oskar Medin, a Swedish pediatrician who, in 1887, recorded the natural history of poliomyelitis and developed a classification that became widely used for decades afterwards. The eponym “Heine-Medin disease” was widely used throughout Europe and North America during the early twentieth century.
Up to this time, only sporadic cases of poliomyelitis were recognized. The first recorded outbreaks of poliomyelitis occurred in the mid- and late nineteenth century in northern Europe and later in North America. Charles Caverly, a Dartmouth College trained physician, described 132 cases with 18 deaths in the Otter Creek Valley near Rutland, Vermont, the largest outbreak known to have occurred up to that time in North America (6). These early outbreaks heralded larger epidemics, including a countrywide epidemic of more than 1,000 cases in Sweden in 1905 (7) and 1,200 cases in New York City in 1907 (1), and the spread of disease throughout Europe and North America. The shift from endemic to epidemic disease has been ascribed to the improved hygienic conditions that accompanied the increasing standards of living in the industrialized world. In theory, extensive exposure to polioviruses during infancy in the presence of passively acquired maternal antibody results in harmless but immunizing infections. Conversely, infections delayed beyond infancy as a result of higher sanitary standards and reduced opportunities for exposure are associated with an increased risk of neurotropic infection (8).
In 1908, Landsteiner and Popper (9) demonstrated that the etiologic agent of poliomyelitis was a “filterable virus” in a series of experiments in which they induced paralysis and spinal cord lesions in a monkey by intraperitoneal injection of neural tissue from a fatal human case; shortly thereafter, Flexner and Lewis (10) at the Rockefeller Institute serially passed polioviruses in monkeys, fulfilling one of Robert Koch’s postulates.
Serologic surveys in the 1930s and 1940s helped define the basis for the observed differences in the behavior of epidemics in different locations and social conditions. Evidence of immunity was found in 80% to 100% of adults tested, with generally lower levels in children (11). In time, it was shown that the risk of infection was universal, but children living in poor sanitary conditions acquired infection at a much earlier age than children from higher socioeconomic backgrounds, an observation that explained the enigmatic shift during the early twentieth century from an endemic disease involving only young infants to an epidemic disease that also affected older children and young adults (12). Later studies conducted by Paul and Riordan (13) in a remote Eskimo population 15 to 20 years following outbreak of poliomyelitis showed that immunity persisted for at least two decades and perhaps for life.
The existence of more than one type of poliovirus was first inferred by Burnet and Macnamara (14) in 1931 when they demonstrated that monkeys who had recovered from infection with a strain recovered in Melbourne subsequently developed disease when given the virulent MV strain and showed that the two strains differed qualitatively in in vitro neutralization tests. These results were confirmed by Paul and Trask (11) at Yale and the two virus groups were respectively designated “Lansing”-like, or “Brunhilde”-like, the latter in reference to the name of a laboratory chimpanzee (1). A third type was predicted by the studies of Bodian (15), at Johns Hopkins in 1949. The existence of three types, and not more, was verified by the testing of poliovirus strains collected worldwide by a collaborative group organized by the National Foundation for Infantile Paralysis (16).
The era of effective poliomyelitis treatment began in the 1920s with the invention of the Drinker negative pressure ventilator or “iron lung” at the Harvard School of Public Health (17) and its development at several Boston hospitals (18). The earliest devices were powered by vacuum cleaners and were fitted with boat portholes for access to the body of the patient. Progress in the management of the persistently paralyzed patient was also made during this period. The practice of rigid immobilization of even mildly involved extremities for long periods gave way to the influential practices of Sister Elizabeth Kenny using warm, moist packs and prolonged physical therapy (19).
In 1936, Sabin and Olitsky (20) produced the first unequivocal evidence that polioviruses could be grown in vitro when they propagated the MV poliovirus strain in human embryonic central nervous system (CNS) tissue, and a decade later, Enders, Weller, and Robbins (21), succeeded in growing Lansing strain poliovirus in human embryonic tissues. This achievement is widely acknowledged as the landmark event that opened the door to further characterization of the virus, better understanding of the pathophysiology of poliomyelitis, and development of successful vaccines.
Building on these advances, Jonas Salk (22) developed the first successful inactivated poliovirus vaccine (IPV), which was first administered to paralyzed patients in a home for crippled children in Pittsburgh in 1952. In 1954 to 1955, Salk IPV was successfully tested in a monumental controlled trial involving more than 1.8 million U.S. schoolchildren. In the meantime, several investigators were studying live, attenuated polioviruses that could be delivered orally and induce mucosal as well as systemic immunity (23). Live attenuated poliovirus strains were tested in humans in several countries and ultimately, three monovalent strains developed by Sabin were licensed for use in the United States in 1961 and 1962. Over the next 2 years, more than 100 million oral poliovirus (OPV) vaccine doses were distributed in the United States via community programs organized by health departments and medical societies. Trivalent OPV vaccine was introduced in 1964 and remained the principal poliovirus vaccine used in the United States until 1997.
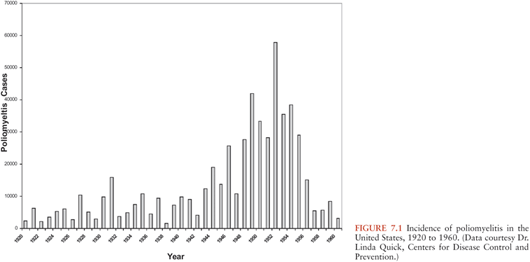
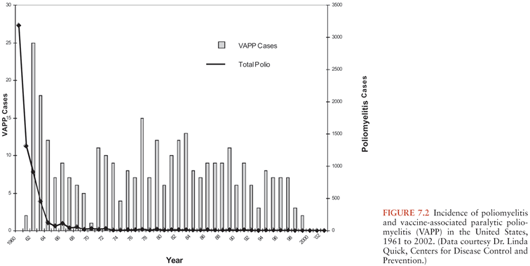
Figure 7.1 illustrates the reported incidence of paralytic poliomyelitis cases in the United States from 1920 to 1960. A peak of 55,000 cases was reported in 1952. The introduction of IPV vaccine for general use in 1955 and OPV vaccine in the early 1960s led to rapid and dramatic control of epidemic poliomyelitis, and the incidence of paralytic poliomyelitis fell from 13.9 cases per 100,000 in 1954 to less than 0.5 cases per 100,000 in 1965. By the 1970s, sustained transmission of naturally occurring polioviruses ceased and the last case of domestically acquired poliomyelitis was reported in 1979 (Fig. 7.2). Rare cases acquired in other countries continued to be imported into the United States in the 1980s, but control of poliomyelitis in the Western Hemisphere by 1991 resulted in virtual disappearance of imported cases. Approximately 8 to 10 vaccine-associated paralytic poliomyelitis (VAPP) cases following administration or transmission of OPV were observed until OPV vaccine was discontinued in favor of IPV in 1997 (24). Since then, only very rare poliomyelitis cases attributed to imported VAPP or to acquisition of OPV vaccine-derived polioviruses (VDPV) by immunodeficient persons have been reported in the United States (25,26).
VIROLOGY, TRANSMISSION, PATHOPHYSIOLOGY, AND IMMUNITY
Virology
The polioviruses are prototypical human enteroviruses (species: Enterovirus C; family Picornaviridae). Three poliovirus serotypes are distinguished from one another by in vitro neutralization with type-specific antisera. The icosahedral virion of approximately 27 nm in diameter is composed of a protein capsid and an enclosed single-stranded RNA genome of approximately 7,500 bases in length. The genome functions as a monocistronic message with a single open reading frame coding a 250 kDa polyprotein, which is subsequently cleaved by both viral coded and cellular proteases into capsid proteins, an RNA polymerase, proteases, and other regulatory proteins. The four capsid proteins (VP1 through VP4) combine to form the 60 structural subunits that are assembled into the viral capsid.
Like all picornaviruses, polioviruses exhibit substantial genomic variability with up to 15% variation within the capsid coding region for each serotype. The three Sabin OPV vaccine strains differ from their naturally occurring parents at fewer than 0.2% of positions across the full genome (27). For all three serotypes, analogous nucleotide substitutions in the 5′ noncoding region appear to be associated with diminished ability to replicate in the gastrointestinal tract and with diminished neurovirulence. Attenuating mutations also map to capsid proteins for each of the individual serotypes.
All three poliovirus serotypes share a common cell membrane receptor (poliovirus receptor [PVR]), a 67-kDa member of the immunoglobulin superfamily coded on human chromosome 19 (28,29). PVR-transgenic mice express PVR in CNS and muscle tissue, endowing susceptibility to these tissues but not on intestinal mucosal cells (30).
Transmission of Polioviruses
Before the introduction of poliovirus vaccines, naturally occurring polioviruses circulated in temperate climates with a marked seasonal variation resulting in peak activity from July to October and low levels from December to May. All three serotypes circulated concomitantly, although one or two types often predominated within a defined community during a single season. Horizontal transmission in the community occurred mostly among children 2 to 5 years of age, and data from virus watch studies indicated that poliovirus transmission was strongly associated with low socioeconomic status and household contact. Once virus was introduced into a household, 90% to 100% of susceptible persons became infected, along with 20% to 50% of seropositive persons (31). Adults were only slightly less likely to become infected than young children. After the introduction of OPV vaccine, the attenuated vaccine viruses replaced the epidemiologic niche inhabited by the naturally occurring viruses. The very low levels of person-to-person transmission of naturally occurring polioviruses during the winter and spring months probably abetted the disappearance of naturally occurring viruses and their replacement by OPV strains (32).
Pathophysiology
Ingested polioviruses implant in the oropharynx and small bowel and penetrate the mucosa via specialized microfold cells (M cells) and other epithelial cells overlying submucosal lymphoid tissues (33). In primate models, the most efficient site of viral replication is detected in Peyer patches in the intestinal submucosa (34,35). Spread to regional lymph nodes gives rise to a transient and clinically silent “minor” viremia during which virus spreads to systemic reticuloendothelial tissue including lymph nodes, bone marrow, liver, and spleen (36). For most poliovirus infections, viral replication is contained at this stage, resulting in subclinical infection. In a minority of infections, further replication of virus in reticuloendothelial tissues leads to a “major” viremia, which coincides with the onset of fever and other clinical symptoms (37).
The path by which polioviruses reach the CNS remains unsettled. It is well established that viremia precedes paralysis, but the precise mechanism by which the virus breaches the blood–brain barrier is not known (38,39). A study using transgenic mice bearing the PVR gene found that spinal cord involvement can be blocked by sciatic nerve section after virus is injected intramuscularly (40), suggesting that virus replication in skeletal muscle precedes transport of virus to the cord via the peripheral nerve (41,42), a concept consistent with the myotropic nature of enteroviruses and the clinical observation of intense myalgias preceding the onset of paralysis in affected patients.
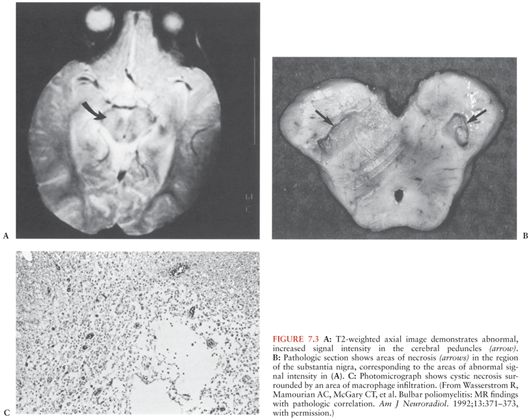
Polioviruses are recoverable from the spinal cord for only the first several days of paralysis. The necrotic lesions and inflammatory infiltrates (Fig. 7.3C), which are distributed within the gray matter of the anterior horn of the spinal cord, the motor nuclei of the pons and medulla, and occasionally other locations (Fig. 7.3A and B), may persist for months (43). The severity of clinical paralysis depends more on the intensity of the lesions, rather than their distribution.
Immunity
Immunity to poliovirus infection is type specific; cross protection between the three serotypes is low, if it exists at all. Reinfection with the same serotype occurs upon exposure to live poliovirus, regardless of whether prior immunity is based on infection with naturally occurring polioviruses, infection with live, attenuated viruses, or inactivated poliovirus vaccine administration. After household exposure to wild type virus, 20% to 50% of naturally immune (31), 30% to 50% of OPV immune, and 90% to 100% of IPV immune persons are reinfected, as defined by virus excretion from the gastrointestinal tract or rise in antibody titer. Reinfections are universally asymptomatic and are rarely associated with oropharyngeal virus excretion.
Passively acquired poliovirus antibody protects against disease, but not against infection (44). Preexisting humoral antibody prevents or reduces oropharyngeal shedding of poliovirus upon reinfection but has only a minor effect on fecal shedding (38,45–47). Serum-neutralizing antibodies are made in response to natural infection and to immunization with either OPV or IPV vaccine. Antibodies made in response to natural infection appear to persist for life (48), and it is presumed that vaccine-induced protection is long term, if not lifelong.
Poliovirus secretory immunoglobulin A (IgA) appears in nasopharyngeal and intestinal secretions 1 to 3 weeks after natural infection or administration of OPV vaccine (49). Secretory IgA antibody persists for at least 5 to 6 years at low levels and, unlike humoral antibody, is not boosted significantly upon rechallenge with OPV vaccine (50). The degree of protection conferred by local IgA antibody is relative. Upon rechallenge, high secretory IgA titers inhibit virus replication, whereas lower titers permit replication; virus shedding is dependent on the challenge dose (46,51). Studies in the field show that childhood recipients of OPV are often reinfected and shed OPV virus in feces despite multiple prior OPV doses (52,53).
CLINICAL FEATURES OF POLIOMYELITIS
Acute Illness
More than 90% of naturally occurring poliovirus infections are inapparent. Acute clinical poliomyelitis is traditionally separated into two distinct phases: the “minor illness” with an incubation period of 3 to 7 days and the “major illness” with onset of symptoms generally 9 to 12 days after exposure (54). The minor illness, coinciding with viremia, consists of nonspecific symptoms such as fever, headache, sore throat, anorexia, and listlessness. Overall, 4% to 8% of infected persons experience symptoms of the minor illness, and most resolve their illness within 1 to 2 days without further symptoms.
The major illness is associated with CNS infection, which has been variously estimated to occur in 0.1% to 1.0% of all poliovirus infections (8,55). About one third of young children who develop the major illness experience a biphasic illness with symptoms of the minor illness preceding onset of CNS disease; adults usually develop CNS disease without the preceding minor illness (37,56). The major illness is heralded by the abrupt onset of fever, headache, vomiting, and meningismus (Table 7.1). CSF pleocytosis is present at this early stage. Approximately one third of cases of CNS disease are limited to meningitis without detectable motor neuron impairment, which resolves within 5 to 10 days (i.e., “nonparalytic poliomyelitis”).
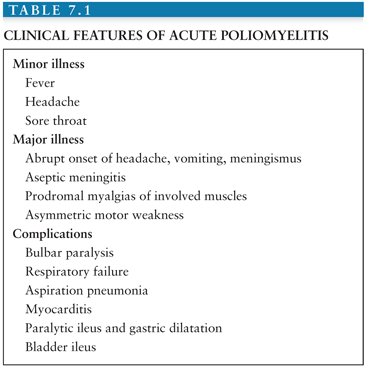
Muscle weakness is preceded by intense myalgias of the involved limb(s) and the axial skeleton. The pain may be relieved by exercise; patients may pace nervously in an attempt to “work off” the pain. The hallmark of poliomyelitis is asymmetric motor paresis, which ranges from mild weakness of a single extremity to complete quadriplegia. Proximal limb muscles are more involved than distal, and legs are more commonly involved than arms. The deep tendon reflexes, which were initially brisk, become absent. The pace of development of the paresis ranges from several hours to several days; most commonly occurring over 2 to 3 days. Cranial nerve involvement (i.e., “bulbar poliomyelitis”) occurs in 5% to 35% of paralytic cases. Any of the motor cranial nerves can be involved, with the ninth and tenth being the most common.
Complications
Bulbar paralysis results in dysphagia, nasal speech, dyspnea, difficulty managing secretions, anxiety, and respiratory compromise. Involvement of respiratory and vasomotor nuclei is less common but may portend serious complications due to hypoventilation, blood pressure lability, and cardiac arrhythmias. Respiratory failure from paralysis of the diaphragm and intercostal muscles represents the most serious complication of paralytic poliomyelitis. Aspiration pneumonia, pulmonary edema, myocarditis, paralytic ileus, gastric dilation, and ileus of the bladder may also complicate acute paralytic disease.
Prognosis
Paralysis most often progresses for 1 to 3 days after onset, rarely more than a week, halting about the time the patient becomes afebrile (57). Most patients with limb paresis experience some recovery of function in the weeks to months after acute disease. An estimate of the eventual outcome can be made by a month after onset when most reversible weakness will have resolved. Very little additional recovery of strength can be expected after a period of 9 months. Residual motor deficits remain in about two thirds of initially paralyzed patients, ranging from minor debility to permanent, flaccid paralysis.
Overall mortality for spinal poliomyelitis is about 4% to 6%. During the poliomyelitis epidemics in the 1940s and 1950s, bulbar polio caused a mortality of 20% to 60% when respiratory assistance was required. With modern intensive care, deaths from temporary respiratory paralysis should be less common.
Differential Diagnosis
Sporadic cases of paralytic disease due to the nonpolio enteroviruses and West Nile virus is clinically indistinguishable from poliomyelitis. Only two nonpolio serotypes have been known to cause epidemic disease: coxsackievirus A7 virus, which has caused small outbreaks of paralytic disease in the former Soviet Union (58), South Africa, and Scotland (59), and enterovirus 71, which has caused large outbreaks in Eastern Europe in the late 1970s and more recently in several countries in Southeast Asia (60–62).
Poliomyelitis must also be distinguished from other causes of acute paralysis including the Guillain-Barré syndrome, which is associated with paralysis that is classically ascending in nature, symmetric, and is accompanied by sensory abnormalities in approximately 80% of cases. The CSF pleocytosis that occurs during the major illness of poliomyelitis is not characteristic of the Guillain-Barré syndrome, which is associated with a normal CSF leukocyte count and an elevated CSF protein concentration. Other conditions produce acute paralysis, including transverse myelitis, botulism, tick paralysis, epidural abscess, cord tumors, and hysteria; however, each of these diseases has features that readily separate them from acute poliomyelitis.
Risk Factors
Although it was once believed that adults are more susceptible to paralytic complications, there is probably little correlation of age with severity of disease after the decline of maternal antibody (8,55). Studies during outbreaks suggest that infected pregnant women have an increased risk of developing paralytic disease (63,64). There is no evidence that either naturally occurring polioviruses (65) or attenuated vaccine polioviruses (66) cause congenital defects.
Persons with B-cell immunodeficiency, primarily young children with X-linked immunodeficiency syndromes, have an increased risk of CNS disease when infected with either naturally occurring or attenuated vaccine polioviruses. These patients may develop acute paralysis or may have an atypical course with an incubation period of several months, prolonged febrile illness, chronic meningitis, and progressive neurologic dysfunction that includes both upper and lower motor neuron involvement (67–70).
Strenuous exercise during the early stages of the major illness substantially increases the risk and severity of poliomyelitis. This effect has been well documented clinically (71,72); and experimental infections with other enteroviruses provide supportive evidence (73).
Paralytic poliomyelitis tends to localize in a limb that has been the site of a recent intramuscular injection or injury (74–76). Observations in experimental poliomyelitis (39,40) and after the administration of OPV vaccine (77) confirm this association but do not fully explain the mechanism of the provoking effect. Tonsillectomy during the incubation period of poliomyelitis markedly increases the risk of bulbar disease (78). The mechanism may be similar to the provoking effect of injections on spinal poliomyelitis.
POSTPOLIOMYELITIS SYNDROME
As many as 20% to 30% of patients who recover from paralytic poliomyelitis experience new onset of muscle weakness, pain, atrophy, and fatigue many years after the acute illness. With the eradication of acute poliomyelitis, the postpolio syndrome remains the sole polio-related clinical disease encountered by physicians in all but a few polio-endemic nations. It is expected that the postpolio syndrome will ultimately decline in incidence and disappear as poliomyelitis is controlled around the globe.
History and Background of the Postpoliomyelitis Syndrome
In 1875, Charcot (79) described a relationship between acute poliomyelitis and late development of motor neuron degeneration. By 1969, approximately 83 cases had been described in which motor neuron degeneration occurred decades after acute poliomyelitis (80), although the distinction between what we now describe as postpolio syndrome and other possible causes of late denervation, such as amyotrophic lateral sclerosis (ALS), was not possible in many of these cases. The hypothesis was advanced that the incidence of late motor neuron disease might increase as children affected during the epidemics of the 1950s reached middle age (81,82). Initial concern that paralytic poliomyelitis might increase the risk of subsequent ALS was allayed by careful epidemiologic studies, suggesting that prior infection with poliomyelitis might actually protect against the subsequent development of ALS (83). At present, it seems clear that the etiology and natural history of the postpolio syndrome and ALS are quite different. Postpolio syndrome is an indolent condition that rarely leads to severe disability or death (84).
Nomenclature
Although the term postpolio syndrome has sometimes been applied to all neurologic symptoms occurring as late manifestations of poliomyelitis, most authorities reserve this designation for a disorder characterized by new-onset muscle weakness, fatigue, and pain, associated with loss of function that occurs years after the original acute poliomyelitis episode (85–87). Another term, postpolio progressive muscular atrophy (PPMA) refers specifically to the weakness with or without atrophy that results from late denervation of muscles of patients (88–91). Criteria for PPMA include a reasonable past history of poliomyelitis with partial recovery of function and at least a decade of stabilization after recovery from the initial illness. PPMA is then the development of progressive muscular weakness following these antecedent criteria. It is worth noting that atrophy probably occurs in only half or fewer of the patients who develop new weakness (92). Although the terms postpolio syndrome and PPMA are used almost interchangeably, it should be recognized that postpolio syndrome or postpoliomyelitis sequelae may include other symptoms such as fatigue (undoubtedly the most common of all symptoms), subjective alterations in attention or cognition, sleep disturbance, and alterations in pain perception. Bulbar symptoms, including dysphagia and some pulmonary disturbances, can occur and may be more common and severe in those who had bulbar symptoms during their childhood illness.
Epidemiology and Risk Factors
Epidemiologic studies indicate that postpolio syndrome affects a substantial proportion of the 250,000 to 640,000 polio survivors estimated to live in the United States (83,86,93). A cohort study of 300 persons living in Olmsted County, Minnesota, who had contracted paralytic poliomyelitis between 1935 and 1955 found that 32 (64%) of 50 poliomyelitis survivors had symptoms consistent with postpolio syndrome, and in another retrospective study of Allegheny County, Pennsylvania, residents with a history of paralytic poliomyelitis noted a prevalence of 28% (94,95). In the latter study, female gender and permanent impairment sustained at the time of acute polio infection seemed to convey a higher risk of developing postpolio syndrome. However, the strongest risk factor for developing postpolio syndrome was the interval from the original paralytic disease with the incidence of postpolio syndrome peaking 30 to 34 years after the original illness. Older age at the time of presentation with postpolio syndrome has also been identified as a risk factor (86).
Etiology
Although the precise cause of postpolio syndrome remains unknown, there is a consensus among authors that the cause is probably related to exhaustion of motor units previously overstressed by recovery from the acute denervation during acute polio virus infection following which terminal elements of surviving alpha motor neurons sprout to reinnervate adjacent myofibrils. After acute poliomyelitis, fewer anterior horn cells innervate a relatively larger number of myofibrils, thereby exaggerating the effects of the additional, physiologic cell loss at a later date attributed to aging (92,96).
Other possible mechanisms have been considered, including immunologic events and other virus–host interactions. Prime among these alternative theories is the conjecture that polioviruses can persist for many years within the CNS of patients with postpolio syndrome and cause recrudescent disease by direct viral infection or immunopathologic pathways (97). Evidence in support of this theory includes the demonstration of oligoclonal immunoglobulin G bands (84,98) or poliovirus-specific immunoglobulin M bands (99) in CSF, and the demonstration of poliovirus-like RNA sequences in CSF of postpolio syndrome patients by the polymerase chain reaction (100). However, the presence of CSF oligoclonal bands has been inconsistent across studies (101,102), and virologists debate the likelihood of infectious virus, or even viral RNA, persisting for long periods in immunocompetent hosts.
Pathophysiology
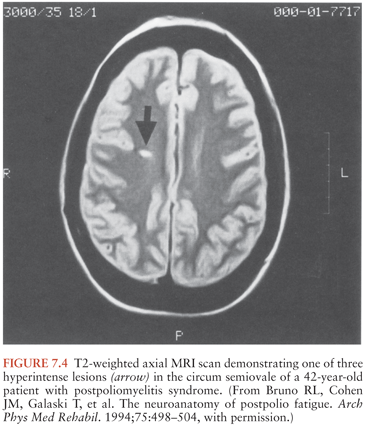
Physical fatigue may result from damage to motor units in previously affected muscle (103–106), perhaps associated with a change in acetylcholine receptor sensitivity and/or number. (This has a practical ramification for anesthesiologists who may find it prudent to reduce the dose of paralytic agents for patients with postpolio syndrome [107].) During exercise, there is decreased maximal voluntary contraction of muscles affected by postpolio syndrome, decreased tetanic force, and delayed recovery. Based on these and other data, Sharma et al. (106) hypothesize that muscle fatigue in postpolio syndrome may be due to impaired activation beyond the muscle membrane at the level of excitation–contraction coupling. Based on neuropathologic studies of patients with poliomyelitis in the 1940s and 1950s, another hypothesis suggests that the fatigue experienced by patients with postpolio syndrome has a central origin. These studies showed lesions in specific areas of the brain whether or not clinical symptoms of encephalitis had occurred, including the reticular formation in the brainstem, vestibular nuclei, cerebellar nuclei, periaqueductal gray, hypothalamic and thalamic nuclei, substantia nigra, locus ceruleus, and median raphe nuclei (43,107,108). The reticular activating system, responsible for maintaining normal alertness and attention, is composed of the brainstem reticular formation, posterior hypothalamus, and thalamus. Specific injuries to these structures from poliovirus infection could cause the acute disorientation and depression of consciousness seen in acute poliomyelitis and hypothetically could set the stage for late-onset decompensation of arousal, attention, and subjective cognition in postpolio syndrome (109,110). It has been suggested that T2-weighted hyperintense areas observed on brain magnetic resonance imaging (MRI) scans could represent these areas of polio virus–induced damage (111) (Fig. 7.4). Injury to the periaqueductal gray of the brainstem and substantia gelatinosa of the posterior horn of the spinal cord may explain the hyperpathia that is reported by many polio survivors (111).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


