HUMAN HERPESVIRUS-6
MARY T. CASERTA
HISTORY
Human herpesvirus-6 (HHV-6) was first isolated from six adults with acquired immunodeficiency syndrome (AIDS) or lymphoproliferative disorders by Salahuddin et al. (1) in 1986. Limited genomic analysis and morphologic studies suggested that the new virus was a member of the herpesvirus family and it was named human B lymphotropic virus (HBLV) based on an apparent B-cell tropism (2). Further experiments demonstrated the propagation of HBLV in various human cell lines, including those of T-cell, B-cell, megakaryocytic, glial, and endothelial origin, and the designation of HHV-6 was proposed (3,4).
In 1988, Yamanishi et al. (5) were the first to report an etiologic role for HHV-6 in human disease. They isolated HHV-6 from the peripheral blood mononuclear cells (PBMCs) of four Japanese children with exanthem subitum (roseola infantum) and demonstrated seroconversion to HHV-6 in the patients’ convalescent sera. Since its discovery, numerous other diseases have been linked with HHV-6 in both immunocompromised and immunocompetent hosts during primary or reactivated infection, but a clear causal connection has not always been proven. Diseases of the central nervous system (CNS) that have been associated with HHV-6 include encephalitis, primarily in immunocompromised hosts, seizures, and multiple sclerosis (MS) (6). Chronic fatigue syndrome, Sjögren syndrome, and several lymphoproliferative disorders including malignancies were all initially associated with HHV-6 but have now been shown by further study to be unlikely due to HHV-6 infection (7–9).
The syndrome of roseola has been well known among pediatricians for generations. Most authors credit Zahorsky (10) in 1913 with the first definite description of the illness as a common, self-limited, febrile exanthematous disease of infancy. In 1941, Breese (11) described the epidemiologic, clinical, and laboratory manifestations of roseola based on his experience with 100 patients. Eight years later, Berenberg, Wright, and Janeway (12) reported convulsions as the most common complication of roseola, occurring in up to one third of children. Kempe et al. (13) were able to document the infectious nature of the illness in 1950 by directly transmitting disease via serum and throat washings from an infected child to an uninfected child and to rhesus monkeys (Macaca mulatta).
EPIDEMIOLOGY
Seroprevalence
Worldwide surveys have demonstrated ubiquitous infection with HHV-6. Seroprevalence rates of more than 80% have been reported consistently in various populations of adults by several methods (14–16). Age-specific studies have revealed that antibody to HHV-6 is universally present in cord blood and newborn sera at titers higher than in maternal sera, with a gradual fall in geometric mean titers and seroprevalence noted in the first 4 months of life, corresponding to the loss of transplacental antibody (17,18). Following this nadir, the geometric mean titers increase steadily to reach a peak at 1 year of age (7,19,20). By 3 years of age, 100% of children have acquired serologic evidence of infection with HHV-6. No seasonal predilection for primary HHV-6 infection has been noted in prospective studies, but small outbreaks of roseola among infants documented by seroconversion to HHV-6 have been reported (21,22). Seroprevalence rates remain stable throughout childhood, adolescence, and adulthood until age 40 years, when a gradual fall in the prevalence and geometric mean titer of antibody to HHV-6 is noted (23,24).
Transmission
The pattern of widespread and rapid acquisition of HHV-6 infection in infancy without contact with similarly ill individuals implies that the virus is spread readily from asymptomatic children and adults to susceptible infants. HHV-6 DNA has been frequently detected via polymerase chain reaction (PCR) in saliva and throat swabs from healthy adults and children both in cross-sectional studies and over time (25,26). Salivary gland tissue specimens screened by in situ hybridization, immunohistochemistry (IHC), and PCR have also been shown to contain HHV-6, suggesting that saliva may be a major mode of transmission and that the salivary glands are a site of persistent infection (27,28). In a prospective, longitudinal study of infants, the risk of acquiring primary HHV-6 infection in the first year of life was greater among subjects whose parents reported saliva-sharing practices, further supporting saliva as an important mode of transmission (29). Recently, HHV-6 DNA has been identified in a majority of nasal mucous and olfactory tissue specimens suggesting that respiratory droplets may also be a source of viral spread (30).
Evidence of primary HHV-6 infection in infants as young as 14 to 21 days documented by viremia and rising antibody titers demonstrates that the protection provided by maternal antibody is incomplete and suggests the possibility of perinatal transmission of HHV-6 (21,31). These data are consistent with reports of HHV-6 DNA in both the blood and the cervical/vaginal samples from 12% to 25.8% of both pregnant and nonpregnant women, implying possible sexual transmission of HHV-6 as well (32–35). HHV-6 DNA was not detected in 120 specimens of breast milk, making breast-feeding an unlikely mode of transmission of HHV-6 (36).
Congenital infection with HHV-6, defined as the presence of viral DNA in cord blood specimens, has been documented in 1.19% (exact CI 1.02, 1.38) of newborns (37). Eighty-six percent of congenital infections are due to chromosomal integration. Transplacental HHV-6 infection accounts for the remaining 14% of congenital infections (38). Chromosomal integration is unique to HHV-6 among the human herpesviruses and has been identified in 0.85% of unselected subjects (39). In individuals with chromosomal integration of HHV-6 (ciHHV-6), the entire viral genome is present in every nucleated cell of the body, including the germline, and transmitted via Mendelian genetics (40). Fluorescent in situ hybridization has demonstrated that multiple chromosomes may harbor integrated virus; however, the viral genome is always identified at the proximal end of the telomere with one site usually identified per family (41,42). The HHV-6 U94 gene product is thought to play a role in HHV-6 latency and in mediating HHV-6 chromosomal integration via homologous recombination between the perfect telomere repeat sequences (TRS) found in the direct repeat segments of the HHV-6 genome and human telomeres (43) (Fig. 13.1).
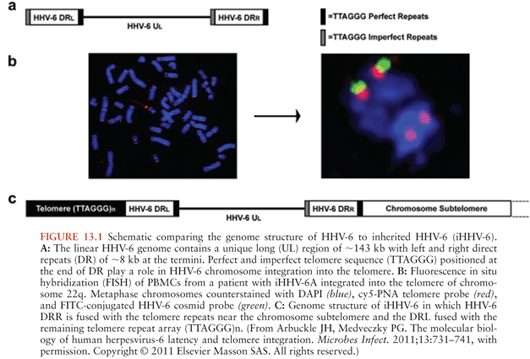
Individuals with ciHHV-6 are notable for having persistently high HHV-6 DNA viral loads in whole blood, serum, plasma, and hair follicle specimens, with one or more genome copies per cell (44). Although replicating virus can be induced from cultured lymphocytes of individuals with ciHHV-6 via treatment with histone deacetylase inhibitors, it is unknown whether the production of infectious viral particles occurs in vivo (41). The observation that infants with transplacental HHV-6 infection have mothers with ciHHV-6 suggests that integrated virus may be able to replicate as free virus and be transmitted (45). No clinical manifestations have been recognized in infants with ciHHV-6 and the long-term clinical significance of chromosomally integrated HHV-6 is unknown. Preliminary data have demonstrated the rate of ciHHV-6 in groups of solid organ transplant patients and those with malignancies to be approximately double that found in the general population suggesting possible pathogenic potential; however, this has yet to be confirmed (39).
Taxonomy
Soon after the initial description of HHV-6, two groups of viruses were identified: HHV-6 variant A and variant B (46). Both were included as members of the Roseolovirus genus in the Betaherpesvirinae subfamily, along with HHV-7 and human cytomegalovirus (CMV) (7). The two HHV-6 variants were found to be highly genetically related but could be distinguished by the pattern of reactivity with monoclonal antibodies, in vitro cellular tropism, nucleotide sequence analysis, and restriction fragment length polymorphisms (7,47,48). In North America, Europe, and Japan, HHV-6B was found in approximately 97% of both healthy and immunocompromised hosts and in infants during primary infection (49,50). Recent PCR data have suggested that HHV-6A is the predominant virus identified in children in sub-Saharan Africa (51). In infants identified with ciHHV-6 in the United States, approximately one third had HHV-6A, a frequency substantially higher than in children with primary infection (21,38). Dual infection with HHV-6A and HHV-6B has also been documented in both adults and children. Because of the differences noted in epidemiology, tissue tropism, genome composition, disease associations, and possible virulence between HHV-6A and HHV-6B, the two were reclassified as separate species in the genus Roseolovirus by the International Committee on Taxonomy of Viruses in 2012. Further clarification of the epidemiology of HHV-6A and HHV-6B awaits the development of reliable species-specific serologic assays (52,53).
VIRAL CHARACTERISTICS
HHV-6 contains a linear, double-stranded DNA genome of 160 to 170 kb that consists of a single unique sequence of approximately 143 kb bounded by a single direct repeat (DR) at both termini. The complete sequences of representative isolates of both HHV-6A and HHV-6B have been published and shown to be approximately 90% identical at the nucleotide level (54–56). The seven conserved gene blocks shared by all HHVs are organized in an identical fashion in HHV-6A, HHV-6B, HHV-7, and CMV, and the unique segment of both the HHV6A and HHV-6B genomes are colinear with the long unique segment of CMV (7). In addition, CMV, HHV-6A, HHV-6B, and HHV-7 share a unique set of genes called beta genes that are found only in CMV, HHV-6A, HHV-6B, and HHV-7 (7).
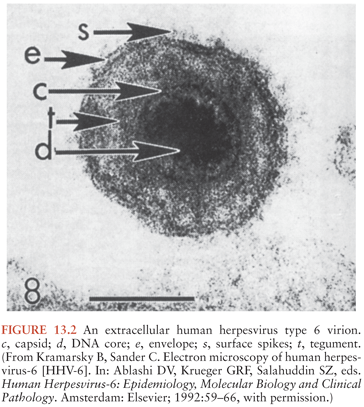
HHV-6 displays the typical ultrastructural characteristics of the herpesvirus family. It is an enveloped virus, approximately 160 to 200 nm in diameter, and contains an electron-dense core. The core is surrounded by a nucleocapsid with icosahedral symmetry with a prominent tegument between the nucleocapsid and outer lipid membrane (57) (Fig. 13.2). HHV-6 viral replication occurs in the nucleus of the infected cell. Viral particles are initially released by exocytosis and then cell death.
Both HHV-6A and HHV-6B have the potential to encode for approximately 97 proteins including those involved in regulatory functions, DNA replication, and structural proteins as well as several unique genes not found in other herpesviruses. Many of these gene products have been identified and reviewed previously and thus are not mentioned here (7,48,58). One notable exception is HHV-6 U94, a protein that is homologous to the parvovirus adeno-associated virus type 2 (AAV-2) Rep 68/78 gene that has been implicated in the maintenance of latency and chromosomal integration as noted earlier (59).
PATHOGENESIS
Viral Growth Characteristics and Cellular Tropism
All HHV-6 isolates can be grown in the laboratory in primary cord blood mononuclear cell cultures that have been pretreated with phytohemagglutinin (PHA) (60). HHV-6 has also been propagated in adult PBMCs that have been stimulated with monoclonal antibody to CD3 but not PHA (61). HHV-6 replication in vitro is augmented by T-cell activation and enhanced in the presence of low concentrations of interleukin-2 (IL-2), with increasing concentrations producing inhibition (62,63). Following infection, HHV-6 reproducibly induces a cytopathic effect characterized by large refractile mononucleated or binucleated cells with intracytoplasmic and/or intranuclear inclusions (64). Infected cells exhibit a slightly prolonged life span in PHA-stimulated cultures compared with uninfected controls; however, lytic infection of mononuclear cells has been the rule (65).
Santoro et al. (66) described CD46 (i.e., membrane cofactor protein) as an essential cellular receptor for HHV-6. CD46 is also the receptor for the Edmonston strain of measles virus and other bacterial microbes and functions as a complement regulatory protein present on the surface of all nucleated cells. This finding is in keeping with the observation that HHV-6 can infect a broad range of cell types in vitro including primary T cells, monocytes, natural killer (NK) cells, dendritic cells, oligodendrocytes, microglia, and astrocytes, with infection of T-cell, B-cell, megakaryocytic, endothelial, glial, and epithelial cell lines described (4,7,60,67–74). Variability has been noted in infectivity between HHV-6A and HHV-6B (69). HHV-6A viruses are most efficiently propagated in the HSB2 or J-Jhan T-cell lines, whereas HHV-6B isolates can be adapted to grow in the Molt-3 T-cell line. A viral glycoprotein complex of gH, gL, gQ1, and gQ2 has been demonstrated to be the viral ligand for many, but not all, laboratory isolates of HHV-6A and HHV-6B (75). Given these observations, the possibility of a second cellular receptor that could explain the differential tropism between HHV-6A and HHV-6B has been postulated. Ablashi et al. (60) studied the kinetics of host cell destruction in T-cell lines and reported that HHV-6A isolates demonstrated enhanced cytopathogenicity compared with HHV-6B isolates suggesting greater virulence of HHV-6A. Despite the broad tropism of HHV-6 noted in vitro, viral replication in nonlymphoid cell lines appears to be substantially restricted with decreased production of infectious virus (58,76).
A previous study of PBMCs from children with roseola suggested that HHV-6 acutely infects primarily mature CD4+ T cells in vivo (77). However, higher viral loads and evidence of viral gene transcription have been demonstrated in monocytes and macrophages compared with CD4+ T cells during the acute phase of primary HHV-6 infection (78). Following primary infection, the HHV-6 genome persists in PBMCs with evidence suggesting that a true state of viral latency is induced in cells of the myeloid lineage including CD34+ stem cells, which may function as a reservoir of latent virus (79,80).
The neuroinvasive potential of HHV-6 has been documented in several reports by the detection of HHV-6 DNA in 32% to 85% of brain tissue specimens from immunocompetent adults at autopsy (81–83). Chan et al. (84) sampled areas of the cerebellum, frontal, temporal, parietal, and occipital lobes bilaterally from 40 patients postmortem and found that 85% had one or more positive specimens. However, there was no association between the presence of HHV-6 DNA and the location of the sample, suggesting that HHV-6 may be widely disseminated in the human brain (84). In addition, HHV-6 DNA has been found in gray matter and white matter samples and in primary brain tumor tissue (82,85). Primary infection with HHV-6 identified by viral isolation from PBMCs is due almost exclusively to HHV-6B. In contrast, HHV-6A DNA was detected in 12% to 70% of human brain tissue specimens in the studies described previously, with up to 21% of patients harboring both HHV-6A and HHV-6B in different regions of the brain (81,82,84,86). In vitro studies have demonstrated HHV-6A and HHV-6B infection of primary astrocytes and oligodendroglia with the formation of multinucleated syncytia (70,87). Attempts to infect neuronal cells have led to conflicting results but generally suggest these cells are not a significant source of productive infection (67,68,87). Enhanced neurovirulence of HHV-6A has been suggested by several in vitro studies examining the degree of cytopathic effect, DNA replication, induction of apoptosis, and inhibition of cell proliferation induced by HHV-6A compared to HHV-6B in both primary glial cells and established cell lines (67,88–91).
The clinical relevance of detecting HHV-6 DNA in cerebrospinal fluid (CSF) samples has not been fully clarified. HHV-6 DNA has been detected in the CSF of one quarter to one half of children with primary infection, indicating that viral DNA may be present in the CSF of a substantial number of immunocompetent children during primary infection (92–94). In one report of young febrile children with evidence of past HHV-6 infection, approximately 30% had detectable HHV-6 DNA present in the CSF (92). The CSF was the only site of viral DNA detection in 29% of children, and these children were significantly younger than those with HHV-6 DNA in both the CSF and the PBMCs, indicating that the CNS may be a preferred site of latency in younger children (92). Typing of CSF samples revealed a greater frequency of HHV-6A in CSF (14%) than in PBMCs (5%), also implying a greater neurotropism of HHV-6A (50). These data imply that finding HHV-6 DNA in the CSF of normal hosts is not a rare event. However, in a single report of bone marrow transplant (BMT) recipients, HHV-6 DNA was identified in 22% of CSF samples from patients with symptoms of CNS dysfunction compared with only 1% of control patients with leukemia (95). Unfortunately, the cases and controls in this study were not well matched by degree of immunosuppression. Nevertheless, these data suggest that detecting HHV-6 DNA in CSF may be a useful marker of disease in the proper clinical setting. Part of the discrepancy between the results from studies of children with primary infection and the BMT patients may be due to the young age of the immunocompetent children studied and the significant percentage with primary HHV-6 infection compared to the older group of BMT patients. Given the recent identification of ciHHV-6 and the fact that most subjects with ciHHV-6 have no symptoms referable to the integrated virus, caution should be used in interpreting the presence of HHV-6 DNA in CSF in order to avoid misdiagnosis of active HHV-6 infection in an individual with ciHHV-6 (39,93). Obtaining HHV-6 viral load measurements in blood and CSF concurrently may be able to help distinguish between these different clinical scenarios (93). Studies of healthy adults have demonstrated the presence of HHV-6 DNA in saliva, PBMCs, urine, and vaginal secretions, with evidence of continued presence of the viral genome at these sites over several months consistent with the broad in vitro tropism of HHV-6 (32,96–98). Despite initial reports of isolation of HHV-6 from adult saliva, subsequent studies have shown that viral isolates obtained from saliva are HHV-7 (99). Additionally, attempts at isolating infectious HHV-6 from healthy subjects’ PBMCs have been generally unsuccessful. In contrast, isolates can be readily obtained from immunocompromised hosts, demonstrating reactivation of latent HHV-6 (100). Reactivation documented either by HHV-6 viral isolation or by quantitative DNA PCR has also been reported in immunocompetent children with measles or dengue hemorrhagic fever and in adults with critical illness requiring intensive care unit care or those with drug induced hypersensitivity syndrome (DIHS) or drug rash with eosinophilia and systemic symptoms (DRESS) (101–104). The cumulative data support the concept of active HHV-6 viral replication in mononuclear cells during primary infection, with the production of abundant infectious particles. The virus then becomes latent or persistent in the host PBMCs, salivary gland tissue, the genitourinary (GU) tract, and the CNS, with the detection of the HHV-6 genome in the absence of significant amounts of infectious virus. Immune dysfunction allows for HHV-6 reactivation, as evidenced by renewed viral isolation with subsequent disease manifestations in a subset of patients.
Immune Response
Children with primary HHV-6 infection develop an immunoglobulin M (IgM) response on day 5 of illness, with the appearance of immunoglobulin G (IgG) at approximately 1 week (105). The IgM response is short lived, persisting for only 2 to 3 weeks, and the IgG response lasts for extended periods. Low level neutralizing antibody develops at approximately day 3 of illness, coincident with a falloff in the rate of viral isolation, and peaks at 2 to 4 weeks into the illness (106,107). Although the immune response to HHV-6 is generally sensitive and specific, IgM has also been detected in approximately 5% of healthy adults and has been shown to be cross reactive with HHV-7, limiting the usefulness of this test for the diagnosis of HHV-6 infection or reactivation (108). HHV-6–specific CD4+ and CD8+ T-cell responses have also been described, supporting a role for cellular factors in the immune response to HHV-6 (109,110).
Following infection of PBMCs, HHV-6 interacts with the immune system in several ways including downregulation of CD3 and CD46 cell surface molecule expression, upregulation of CD4, induction and suppression of cytokines and type I interferons, and effects on NK cell activity (107,111–114). In vitro HHV-6 infection of dendritic cells has been described with conflicting reports on the functional outcome of this infection (115,116). Apoptosis of T cells both in vitro and in vivo has been detected following HHV-6 infection, as well as the previously well-described cell lysis (117,118). In addition to the immune response generated to HHV-6, the viral genome itself encodes several factors that have immunologic regulatory activity including two chemokine receptor homologs (U12 and U51) and two putative chemokines (U22 and U83) (111). It has been postulated that through these proteins, HHV-6 may either enhance viral replication in infected cells, enlarge the pool of available virus susceptible cells, or circumvent the host immune response to infection.
CLINICAL MANIFESTATIONS
Primary Infection
Roseola is an acute self-limited disease of infancy and childhood. It is characterized by the abrupt onset of high fever lasting approximately 72 hours. The fever resolves by crisis coincident with the appearance of a faint morbilliform rash on the neck and trunk. Associated signs are notably sparse but can include mild injection of the pharynx, palpebral conjunctivae, or tympanic membranes and enlarged suboccipital nodes (11).
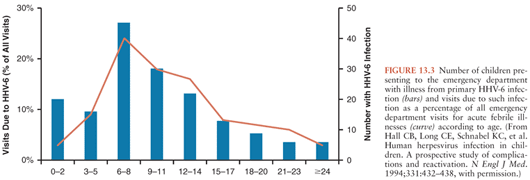
Prospective studies have extended our knowledge of the clinical manifestations of acute HHV-6 infection beyond that of classic roseola. Hall et al. (21) identified 160 children with primary HHV-6 infection by viremia and seroconversion from a large number of children evaluated in a pediatric emergency department. The mean age at primary HHV-6 infection was 9.4 months, with few cases occurring before 2 months or after 25 months of age (Fig. 13.3). High fever was the most consistent manifestation of infection. All children had an acute febrile illness, and 87% had fever of more than 39°C. Fever persisted for 6 days or longer in 15%. Associated clinical symptoms and signs included irritability or a toxic appearance in 72%, inflamed tympanic membranes in 46%, and upper respiratory tract signs in 41%. Children with primary HHV-6 infection were significantly more likely to exhibit these signs than matched controls. Cough and lower respiratory tract signs were seen significantly less often in children with primary HHV-6 infection than controls. Only 17% of children with primary HHV-6 infection developed a rash, making it difficult to identify correctly the cause of the illness. Most children were given a diagnosis of fever with otitis media, fever of undetermined origin, or presumed sepsis, and 13% were hospitalized. A more recent prospective study of an outpatient cohort of infants with primary HHV-6 infection identified by HHV-6 DNA detection in saliva confirmed that over 90% of the infants had symptoms with fever, fussiness, and rhinorrhea noted most often (119). Rash was identified in a little less than one third of the cohort (119).
In a report from Japan of 176 children with roseola identified retrospectively, fever and rash were noted in 98% of the patients (120). This study also detected a higher percentage of children with gastrointestinal tract disturbances (68% with diarrhea), in addition to edematous eyelids in 30%, erythematous papules in the pharynx in 65%, cough in 50%, and mild cervical lymphadenopathy in 31%. The differences between these studies may be attributed in part to study design and case definition. Also, the severity of the clinical symptoms associated with acute HHV-6 infection has been shown to be related to the degree of viremia, which may contribute to the range of reported manifestations of primary HHV-6 infection (121).
The laboratory evaluation of children with primary HHV-6 infection is generally nonspecific and reveals a depressed total white blood cell count, with lymphopenia noted early in the course of the infection. C-reactive protein levels have been reported to be less than 10 mg/L in most patients (21,122). Rarely reported complications of primary HHV-6 infection include intussusception, Kawasaki disease, hepatitis, hemophagocytic syndrome, and fatal multisystem disease (7,123). A heterophil-negative mononucleosis syndrome has been associated with primary HHV-6 infection in immunocompetent adults (124). However, most reports have relied on seroconversion as evidence of HHV-6 infection, a method that is not optimal.
Central Nervous System Complications of Primary Infection
Seizures have been reported as a complication of primary HHV-6 infection. In the study from Japan noted earlier, 8% of the children with roseola had seizures during the febrile phase of the illness (120). Seizures were the most common complication of primary HHV-6 infection, occurring in 13% of children with acute infection described by Hall et al. (21) from a pediatric emergency department population. The highest incidence of seizures was 36%, noted in children 12 to 15 months of age. This rate was significantly greater than that of age-matched controls with other febrile illnesses. In prospective studies of children with first time febrile seizures, primary infection with HHV-6 has been observed in 18% to 26% of the patients (125,126). However, in the study by Zerr et al. (119) noted earlier, no seizures were recorded among 81 ambulatory children with primary HHV-6 infection. Although the exact rate of seizures due to primary infection with HHV-6 has not been precisely defined, a review of the viral causes of febrile seizures examined almost 1,000 published cases of primary HHV-6 infection and found seizures to be a complication in 16.5% (127).
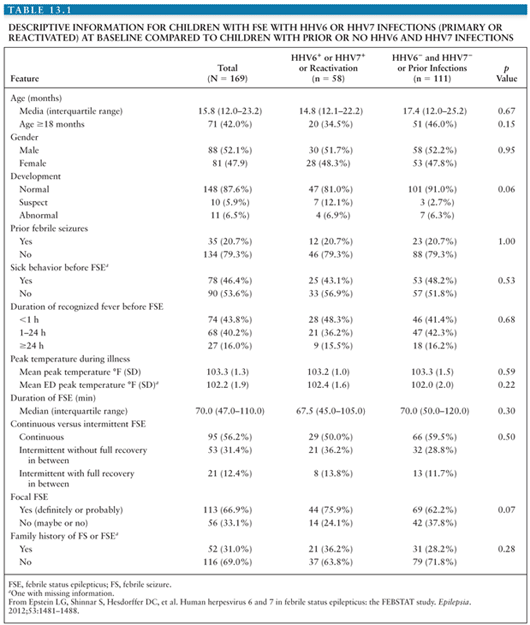
Clinically, no differences have been identified between children with febrile seizures associated with primary HHV-6 infection and those with febrile seizures not associated with primary HHV-6 infection, including the height or duration of fever or the recurrence rate of seizures in the first 24 hours (125). Additionally, CSF analysis does not typically reveal pleocytosis or abnormal glucose or protein levels in patients with primary HHV-6 infection. Suga et al. (128) reported a higher frequency of partial seizures, postictal paralysis, prolonged seizures, and clustering of seizures in 19 children with primary HHV-6 infection compared with children with seizures not associated with primary HHV-6 infection, suggesting more significant CNS pathology and implying an increased risk for the later development of epilepsy (92,129). Early reports did suggest an increased risk of recurrent febrile seizures in children due to HHV-6 infection, but a case–control study of children with febrile seizures associated with primary HHV-6 infection compared with children with febrile seizures not associated with primary HHV-6 infection demonstrated a statistically significant lower rate of seizures over the following 3 years in the children with primary HHV-6, casting doubt on this association (130). However, more recent data have suggested a link between chronic smoldering or reactivated HHV-6 infection of the CNS and the later development of medial temporal lobe epilepsy (MTLE) (131). One line of evidence supporting this association comes from a prospective study of children with febrile status epilepticus (FSE), a known risk factor for the development of MTLE that identified active HHV-6 infection in peripheral blood samples of almost one third of the patients (132). Similar to the data from children with simple febrile seizures, the children with FSE associated with HHV-6 did not have CSF pleocytosis or differences in peripheral white blood cell counts from patients with FSE not associated with HHV-6. There were also no differences in the proportion of children with EEG abnormalities or acute hippocampal changes on MRI. Seizure characteristics between children with FSE associated with HHV-6 and those not associated with HHV-6 were similar (Table 13.1), suggesting a shared pathogenesis of FSE among different etiologies (132).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


