INTRODUCTION
SUMMARY
The safe and effective use of anticancer drugs in the treatment of hematologic malignancies requires an in-depth knowledge of the pharmacology of these agents. In this field of medicine, the margin of safety is narrow and the potential for serious toxicity is real. At the same time, anticancer drugs cure many hematologic malignancies and provide palliation for others. The discovery and development of treatments for leukemia and lymphoma have provided a paradigm for approaches to the improved treatment of the more common solid tumors.
The intelligent use of these drugs begins with an understanding of their mechanism of action. Cytotoxic anticancer drugs inhibit the synthesis of DNA or directly attack its integrity through the formation of DNA adducts or enzyme-mediated breaks. These DNA-directed actions are recognized by repair processes and by the checkpoints that monitor DNA integrity, including most prominently p53. If DNA damage cannot be repaired, and if the DNA damage reaches thresholds for activating programmed cell death, then DNA damage is translated into tumor regression. Attention has turned to the possibility of identifying molecular targets unique to tumor cells, or dramatically overexpressed in those cells, including molecules involved in cell signaling and cell-cycle control, but the principles of drug action and resistance to these compounds remain the same. Resistance to drug action can arise from alterations in any one of the critical steps required for drug activity; these steps include drug uptake and distribution through the bloodstream or across the blood–brain barrier; transport across the cell membrane; transformation of the parent drug to its active form within the tumor cell or in the liver; interaction of the drug with its target protein or nucleic acid; enzymatic or chemical inactivation of the agent; drug transport out of the cell; and elimination of the agent from the body through the kidneys or through metabolic transformation. The underlying mutability of tumors leads to the spontaneous generation of cells with alterations in drug uptake, transformation, inactivation, and target binding. In the presence of the selective pressure of drug, resistant tumors replace sensitive cells as the dominant tumor population. Combination chemotherapy overcomes resistance that carries specificity for single agents, but the expression of multidrug resistance genes, as well as loss of the apoptotic response, can result in resistance even to combination drug therapy.
In addition to the molecular determinants of drug action, pharmacokinetics (the disposition of drugs in humans) plays a critical role in determining drug effectiveness and toxicity. Drug regimens are designed to achieve a maximally effective concentration in plasma and tumor cells for an effective duration of exposure. Because of the potential of these agents for toxicity, it is critical for hematologists and oncologists to understand the pathways of drug clearance and to adjust dose in the presence of compromised organ function. Drugs such as methotrexate, hydroxyurea, and the newer purine antagonists (fludarabine and cladribine) are eliminated primarily by renal excretion and should not be used in full doses in patients with renal dysfunction. Similarly, hepatic dysfunction with an elevated serum bilirubin concentration should alert clinicians to decrease doses of the taxanes, vinca alkaloids, and the anthracyclines. In addition, clinicians must be alert to the potential for drug interactions, particularly the ability of drugs that induce or inhibit cytochrome metabolism to alter patterns of drug elimination.
Inherited genetic variations in drug-metabolizing enzymes may lead to an increased risk of drug toxicity and may alter the antitumor response. The most important of these familial syndromes affecting treatment of leukemia is the deficiency of thiopurine methyltransferase, which slows the elimination of 6-mercaptopurine (6-MP) and leads to unanticipated toxicity during maintenance chemotherapy for acute lymphocytic leukemia. Pharmacokinetic monitoring has a standard role in the use of certain therapies, particularly high-dose methotrexate, and in the evaluation of new drugs or new drug combinations. To ensure appropriate dosing, and management of toxicity, there is no substitute for therapy based on standard protocols and peer-reviewed clinical trials. Adherence to protocols ensures that the pharmacologic variables affecting drug disposition can be taken into account early in the course of treatment and that serious untoward events can be avoided while maintaining effective therapy.
Acronyms and Abbreviations:
ABVD, Adriamycin (doxorubicin), bleomycin, vinblastine, and dacarbazine; ADCC, antibody-dependent cellular cytotoxicity; αKG, α-ketoglutarate; ALL, acute lymphocytic leukemia; AML, acute myelogenous leukemia; APL, acute promyelocytic leukemia; ara-C, cytarabine; ara-CTP, cytarabine triphosphate; ara-G, arabinosylguanine; ara-GTP, arabinosylguanine triphosphate; ara-U, arabinosyluracil; ATRA, all-trans retinoic acid; BCNU, bischloroethylnitrosourea; BCRP, breast cancer resistance protein transporter; BET, bromodomain and extraterminal; BTK, Bruton tyrosine kinase; CHF, congestive heart failure; CLL, chronic lymphocytic leukemia; CML, chronic myelogenous leukemia; COMFORT, Controlled Myelofibrosis Study with Oral JAK Inhibitor Treatment; CrCl, creatinine clearance; CYP, cytochrome P450; dCK, deoxycytidine kinase; dCTP, deoxycytidine triphosphate; DHFR, dihydrofolate reductase; DNMT, DNA methyltransferase; DTIC, dimethyltriazenoimidazole carboxamide; EPO, erythropoietin; ET, essential thrombocythemia; etoposide, VP-16; EZH2, enhancer of zest homologue 2; FBP, folate-binding protein; GM-CSF, granulocyte-macrophage colony-stimulating factor; HDAC, histone deacetylase; hENT, human equilibrative nucleoside transporter; 2HG, 2-hydroxyglutarate; Hgb, hemoglobin; HGPRT, hypoxanthine guanine phosphoribosyltransferase; IC50, inhibiting growth by concentration 50 percent; IDH, isocitrate dehydrogenase; IL, interleukin; IMiD, immunomodulatory drug; IRF4, interferon regulatory factor 4; IRIS, International Randomized Study of Interferon and STI571; JAK, Janus-type tyrosine kinase; JMJC, Jumonji-C domain; MDR, multidrug resistance; MDS, myelodysplastic syndrome; mesna, sodium 2-mercaptoethane sulfonate; MLL, mixed-lineage leukemia; MOPP, nitrogen mustard, vincristine (Oncovin), procarbazine, and prednisone; 6-MP, 6-mercaptopurine; MPN, myeloproliferative neoplasm; MRI, magnetic resonance imaging; MRP, multidrug resistance-associated protein; MTD, maximum tolerated dose; MUGA, multigated acquisition scan; NK, natural killer; OCT1, organic cation transporter-1; PDGFR, platelet-derived growth factor receptor; PEG, monomethoxypolyethylene glycol; PMF, primary myelofibrosis; PRC2, polycomb repressive complex 2; PRPP, phosphoribosyl pyrophosphate; PV, polycythemia vera; RARα, retinoic acid receptor-α; REMS, risk evaluation and mitigation strategy; RNR, ribonucleotide reductase; SAMe, S-adenosyl-L-methionine; S-phase, synthetic-phase; STAT, signal transducer and activator of transcription; teniposide, VM-26; 6-TG, 6-thioguanine; TKI, tyrosine kinase inhibitor; Topo II, topoisomerase II; TPMT, 5-thiopurine-methyltransferase; TPO, thrombopoietin.
The leukemias and lymphomas have been the initial testing ground for cancer chemotherapy. Because of their rapid rates of proliferation, lack of surgical treatment options, ready access to malignant cells, and availability of mouse models of leukemia, the hematologic malignancies drew the attention of early investigators interested in treating cancer with drugs. The first evidence for activity of a chemical antitumor agent came in 1942, from the experimental work and subsequent clinical trials conducted by Goodman, Gilman, and colleagues at Yale, and their observation that nitrogen mustard caused tumor regression in a patient with Hodgkin lymphoma.1 Six years later, Sidney Farber, a pathologist at Children’s Hospital in Boston, made the even more startling discovery of remission induction by aminopterin and then methotrexate in acute lymphocytic leukemia (ALL). His work ushered in the modern era of chemotherapy.2 Over the next 20 years, clinical trials in these diseases established the basic principles of cyclic combination therapy and dose intensification,3 developed effective strategies for management of infectious and hemorrhagic complications, and led to the cure of these diseases with chemotherapy. High-dose chemotherapy with marrow reconstitution has further extended the cure rate in leukemias and lymphomas. As our understanding of the biologic and molecular basis for malignancy has advanced, the concept of molecularly targeted therapy achieved its first striking success with the development of imatinib mesylate for chronic myelogenous leukemia (CML).4 Studies of relapsing patients on imatinib provided the first clear evidence for target mutation as a mechanism of clinical drug resistance.5 The first effective use of a monoclonal antibody, rituximab, has extended the cure rate for patients with large cell lymphomas, and the first clear demonstration of drug-induced differentiation by all-trans retinoic acid (ATRA) has led to a remarkable improvement in the cure rate of acute promyelocytic leukemia (APL).6 Other unique noncytotoxic drugs with unusual mechanisms of action, such as L-asparaginase, thalidomide, and bortezomib, have become valuable components of regimens for specific kinds of hematologic malignancies. Molecular studies of the abnormalities in pathways that control proliferation and survival lymphomas and leukemias have revealed distinct subsets of disease have identified new therapeutic targets.
BASIC PRINCIPLES OF CANCER CHEMOTHERAPY
The safe and effective use of chemotherapy in clinical practice requires a thorough understanding of the basic aspects of drug action as well as knowledge of the important clinical toxicities, pharmacokinetics, and drug interactions of the various agents. Antineoplastic chemotherapy is a complex undertaking, with the potential for serious or fatal side effects. Patients are best served if their treatment is based on evidence from clinical trials, which define optimal doses, schedules, and drug combinations. The specific protocol chosen for treatment should be appropriate not only for the stage and histology of the tumor but should consider individual patient comorbidities, age, and susceptibility to specific potential toxicities. Thus, bleomycin is not a safe choice for a patient with serious underlying renal or lung disease, nor is doxorubicin an appropriate drug for use in a patient with a history of congestive heart failure; even in patients with normal cardiac or pulmonary function, total dose limits should be respected for these agents. Even though clinical trials define the benefits and risks of a cohort of patients of a defined age range and physiology, these results may not be easily extrapolated to patients at the extreme ends of the spectrum.
Depending on the major route of drug clearance, doses should be modified for renal or hepatic dysfunction (Table 22–1). Changes in the dose and schedule of a drug (dose-dense chemotherapy) offer potentially greater antitumor effects, but often lead to unique toxicities. With the development of techniques for marrow or blood stem cell storage and replacement of marrow after chemotherapy, potentially lethal doses of chemotherapy can be administered in an attempt to cure malignancies refractory to standard regimens. In general, these regimens may produce organ toxicities not seen at conventional doses—including pneumonitis, cardiac failure, vascular endothelial damage, and hepatic and renal insufficiency—and are ordinarily reserved for patients of younger age and with normal baseline organ function.
Renal dysfunction (creatinine clearance <60 mL/min) Reduce dose in proportion to reduction in creatinine clearance. Drugs
Hepatic dysfunction For bilirubin >1.5 mg/dL reduce initial dose by 50%. For bilirubin >3.0 mg/dL reduce initial dose by 75%. Drugs
|
The success of chemotherapy in curing hematologic malignancy is incompletely understood. Although targeted therapies exploit clear differences in biology conferred by mutations or amplification of key genes, an explanation for the differential effects of cytotoxic drugs on tumor versus normal tissues is less obvious. The greater susceptibility of malignant cells to drug toxicity, as reflected in the phenomenon of leukemia remission induction, with restoration of normal marrow function, may result from the relative resistance of normal marrow stem cells to drug injury. These stem cells exist in a nonreplicating phase of the cell cycle, where they are less susceptible to damage by DNA-directed agents, and they express genes that protect against chemical and hypoxic damage. In addition, there is growing evidence that cancer cells lack cell-cycle checkpoints that recognize DNA damage and activate repair of DNA strand breaks, base deletions, or other lesions induced by chemotherapy. This differential in repair capability may allow normal cells to repair damage and promote recovery from chemotherapy-induced injury.
Most leukemias and lymphomas are highly drug sensitive, but, with the exception of the curability of Burkitt lymphoma (treated with cyclophosphamide) and hairy cell leukemia (treated with cladribine), are rarely, if ever, cured with single-agent chemotherapy. Combination chemotherapy forestalls the emergence of drug-resistant cells and thus is curative in settings where individual agents are ineffective. Empiric principles have resulted from the clinical experience of the past 4 decades of combination therapy. In general, drugs selected for combination therapy should have demonstrable antineoplastic activity, or at least biologic effects, against the tumor in question. The lone exception may be targeted drugs that inhibit signal transduction or angiogenesis; antibodies such as trastuzumab or rituximab may exhibit limited antitumor activity on their own, but may significantly augment the action of cytotoxic agents.7 Individual agents in a combination should be chosen based on their different mechanisms of action and lack of a common mechanism of resistance such as multidrug resistance (MDR). The dose-limiting toxicities of the agents chosen should not overlap; otherwise, they could not be used together at or near full doses. The clinical use of specific combinations should be based on preclinical evidence of synergistic interaction, and single-agent activity in the disease in question. Favorable molecular or biochemical drug interactions may be dependent on specific schedules of administration. Pharmacokinetic interactions should be defined in initial trials of drug combinations so as to avoid under- or overdosing of individual agents.
Another important consideration in designing clinical protocols is dose intensity, the dose administered per unit time, which should be maintained throughout a treatment regimen. Achieving this objective may require the use of hematopoietic growth factors to hasten marrow recovery, prevent repeated episodes of febrile neutropenia, and allow ontime administration of the next treatment cycle.
Interdigitation of chemotherapy with surgery and irradiation makes it possible to take advantage of favorable cytokinetic or radiosensitizing effects of chemotherapy, but drugs may enhance radiation or surgical toxicity to normal organs, an interaction that requires careful consideration in designing a multidisciplinary regimen. Thus, 5-fluorouracil and cisplatin are potent radiosensitizers used with radiation therapy to enhance local tumor control in solid tumors. In the treatment of lymphomas, the toxicity of radiation therapy to sensitive organs such as skin, lung, heart, and brain may be significantly increased by concurrent administration of anthracyclines, a consideration that has prompted the use of radiation therapy either before or after anthracycline antibiotics, but not concurrently. Likewise, bleomycin sensitizes the lungs to damage by high concentration of inspired O2 during surgery. Antiangiogenic therapies, rarely used in hematologic malignancies, are associated with an increased risk of bowel perforation in patients who have recently undergone intraabdominal surgery.
The cell-killing characteristics of cancer chemotherapeutic agents vary according to their mechanism of action. Many of the most effective agents in antileukemic therapy belong to the antimetabolite class, including cytarabine and methotrexate. These drugs kill cells most effectively during the DNA synthetic phase (S-phase) of the cell cycle. For these agents, a prolonged period of tumor exposure to drug is essential so as to maximize the number of cells exposed during the vulnerable period of the cell cycle. As would be predicted, the antimetabolite drugs are primarily active against rapidly dividing tumors such as acute leukemias and intermediate and high-grade lymphomas. Other anticancer drugs, such as the topoisomerase inhibitors and alkylating agents, do not require cells to be exposed during a specific phase of the cell cycle, although like the antimetabolites, these drugs are generally more effective against actively proliferating cells as compared to resting cells. Still others, most notably the nitrosoureas and busulfan, are equally toxic to dividing and nondividing cells, and at the same time, deplete marrow stem cells. In general, the toxicity of alkylating agents is determined by the total dose of drug, whereas for the cell-cycle-specific drugs (such as methotrexate and cytarabine), both drug concentration and duration of exposure determine cytocidal effect. However, for drugs that act through alternate mechanisms, such as the taxanes, myelosuppression correlates best with the duration of exposure above a threshold plasma concentration, which is approximately 50 to 100 nM for paclitaxel and 200 nM for docetaxel.8
High-dose regimens achieve a number of worthwhile objectives for these agents, including an enhancement of cross-membrane transport, saturation of anabolic pathways inside the cell, and prolongation of the period of effective drug concentration. However, achieving these objectives is realized at the cost of increased toxicity to normal proliferating marrow precursor cells and may produce significant and unexpected damage to normal organs, such as hepatic venoocclusive disease (alkylating agents), cerebellar toxicity (certain alkylating agents and cytarabine [ara-C]), or pulmonary toxicity (nitrosoureas and alkylating agents). Because hematopoietic stem cells can be harvested, stored, and reinfused, dose-limiting toxicities of high-dose chemotherapy are generally those affecting nonhematologic organs.
The choice of an appropriate dose and schedule of drug administration depends on a number of factors: (1) the drug’s cell-cycle dependence; (2) the often empirically derived relationship between antitumor effects, drug dose, and schedule; (3) pharmacokinetic behavior and the need to maintain a specific drug concentration for a given period of time; (4) potential interactions with other components of the treatment regimen; and (5) patient tolerance. Multiple clinical trials are required to establish safe and effective single-agent regimens and drug combinations. For molecularly targeted drugs, the aim is to maintain inhibition of the target for prolonged periods of time, keeping drug levels above a threshold for toxicity to tumor cells, but balancing these considerations against the potential for toxicity to normal tissues, such as skin, liver, and intestinal epithelium.
Inadequate treatment of a sensitive tumor tends to select for the outgrowth of drug-resistant clones of the original tumor. The reasons for emergence of drug resistance are manifold. Cancer cells often harbor basic defects in DNA repair as one of their hallmark mutations and spontaneously generate drug-resistant mutants, even in the absence of drug exposure. Thus it has been demonstrated in the specific example of imatinib treatment of CML that drug-resistant cells, carrying specific mutations in the BCR-ABL gene, can be identified in marrow prior to treatment and become the dominant tumor population under the selective pressure of drug treatment.5 A similar finding of pretreatment mutations explains drug resistance to inhibitors to the epidermal growth factor receptor in non–small-cell lung cancer.9 In addition, many cancer drugs, especially alkylating agents, and irradiation are mutagenic and increase the rate of generation of drug-resistant mutants, as demonstrated in the selection of mismatch repair mutants by temozolomide.10 To discourage the outgrowth of resistant cells, multiple agents with differing mechanisms of resistance should be used simultaneously, because the likelihood of there being a doubly or triply resistant cell is the product of the probabilities of the independent drug-resistant mutations occurring at the same time in a single cell. The probability of a cell division resulting in mutation at any given genetic locus is approximately 10–6 for any given episode of cell division in somatic cells; thus the probability of two independent mutations arising in the same cell is 10–12. Mutation rates may be distinctly higher in tumor cells and may be further increased by exposure to alkylating agents and irradiation. Some mutations, such as those affecting apoptosis, may confer resistance to multiple agents of diverse mechanisms of action. Thus, the probability of encountering MDR cells is much higher in reality.
In choosing drugs for combination therapy, one must bear in mind potential mechanisms of resistance. Classical MDR occurs as a consequence of increased expression of drug efflux pumps such as the P-glycoprotein or the MDR-associated proteins (MRPs),11,12 and confers resistance to a broad spectrum of agents derived from natural products, including taxanes, anthracyclines, vinca alkaloids, and epipodophyllotoxins, and potentially to a number of “targeted” agents. Other mechanisms of resistance that induce amplification of a target gene, such as dihydrofolate reductase (DHFR)13 or BCR-ABL kinase,14 may be highly specific for a single drug. Table 22–2 lists the common mechanisms of resistance. Although the presence of these biochemical changes is not routinely determined in tumor biopsies prior to therapy, these mechanisms should be considered in developing new protocols and in choosing cytotoxic therapy. In relapse after targeted therapy of CML or certain non–small cell lung cancers, the choice of second-line therapies may rely on studies of tumor cell resistance, as reflected in repeat biopsies of solid tumors or cell sampling in CML.
| Mechanisms | Drugs Affected | Clinical Role |
|---|---|---|
1. Decreased drug uptake Reduced folate transporter Nucleoside transporter | Methotrexate cytarabine | ALL AML |
2. Increased drug efflux MDR transporter (P-glycoprotein) MRP transporters, breast cancer- resistant protein | Anthracyclines, vinca alkaloids, taxanes, etoposide Anthracyclines, vinca alkaloids, taxanes, etoposide | Myeloma, AML, non-Hodgkin lymphoma Breast cancer |
3. Decreased drug activation in tumor Deoxycytidine kinase deletion Hypoxanthine phosphoribosyltransferase deletion Folylpolyglutamation | cytarabine, fludarabine, cladribine, clofarabine 6-Mercaptopurine Methotrexate | AML, CLL, hairy cell leukemia Uncertain Acute leukemias |
4. Increased drug inactivation defect Thiopurine methyltransferase Bleomycin hydrolase Glutathione transferase | 6-Mercaptopurine Bleomycin Alkylating agents | ALL Uncertain Uncertain |
5. Decreased target enzyme Topoisomerase I Topoisomerase II | Camptothecins Anthracyclines, etoposide | Uncertain Uncertain |
6. Increased target enzyme Dihydrofolate reductase Thymidylate synthase Adenosine deaminase | Methotrexate 5-Fluorouracil Deoxycoformycin | Acute leukemia, small cell lung cancer Solid tumors Lymphoid tumors |
7. Mutated intracellular target BCR-ABL kinase Tubulin Topoisomerase I Topoisomerase II | Imatinib mesylate, dasatinib Vinca alkaloids, taxanes Camptothecins Anthracyclines, etoposide | CML Uncertain Uncertain Uncertain |
8. Increase DNA repair Guanine-O-6-methyltransferase Nucleotide excision repair | Procarbazine, nitrosoureas temozolomide Platinating drugs | Brain tumors Ovarian cancer |
9. Decreased DNA damage recognition p53 mutation Mismatch DNA repair mutations | Many cancer drugs, radiation Platinating agents, methylating drugs, thiopurines | Leukemias, lymphomas Colon cancer, glioblastoma, leukemias |
In addition to drug-specific mechanisms of resistance, mutations that abolish recognition of DNA damage, such as the loss of components of the mismatch repair gene complex (MLH6 or MSH2)15 seem to block initiation of apoptosis by cisplatin, thiopurines, or alkylating agents. Other mutations that block the induction of apoptosis, such as loss of p5316 or overexpression of the antiapoptotic factors such as BCL-2,17 may render tumor cells insensitive to a broad array of drugs and modalities, including ionizing irradiation, alkylating agents, antimetabolites, and anthracyclines. Although the specific contribution of p53 mutation and altered apoptosis to clinical resistance is still uncertain, emerging evidence suggests that these factors are commonly associated with clinical resistance and aggressive tumor growth and may be more relevant causes of drug resistance in the clinic than are the classical drug-specific mechanisms found in experimental tumors.
The contribution of tumor stem cells to treatment resistance and disease recurrence is an intriguing, but as yet undefined, possibility. It is clear that many tissues, including marrow, contain stem cells capable of repopulating organs, even from single cells.18 Likewise, many tumors contain stem cells, which, on careful evaluation, preserve many of the surface antigens of their normal counterpart, and display resistance to DNA damage, reactive oxygen species generated by drugs or irradiation, and readily export toxic natural products.19 It is possible, but still to be established, that these drug-resistant stem cells represent the ultimate barrier to successful cancer treatment.
CELL-CYCLE-SPECIFIC AGENTS
A number of anticancer drugs, particularly those developed during the era of cytotoxic chemotherapy, exert their antitumor effects on DNA synthesis. Cells are thus most vulnerable during periods of active DNA synthesis (S-phase), and least affected during quiescent (G0) stages of their life cycle. Thus tumors that have a high proliferative rate, such as leukemias and aggressive lymphomas, are most vulnerable to these agents.
Farber and associates showed that the folate antagonist aminopterin induced a complete remission in children with ALL, thereby launching the modern era of chemotherapy. Unfortunately, these remissions were short-lived, and the leukemia invariably became resistant to further treatment. Subsequently, methotrexate, a 4-amino, N-10 methyl analogue of folic acid, supplanted aminopterin because it had more predictable side effects. Methotrexate continues to be a key drug in the induction and maintenance therapy of ALL, in the intrathecal prophylaxis and treatment of CNS leukemia, in the primary treatment of CNS lymphomas, and in combination therapy of high-grade lymphomas.
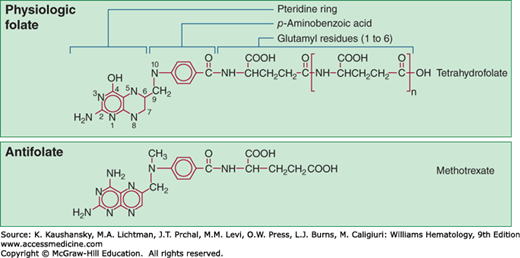
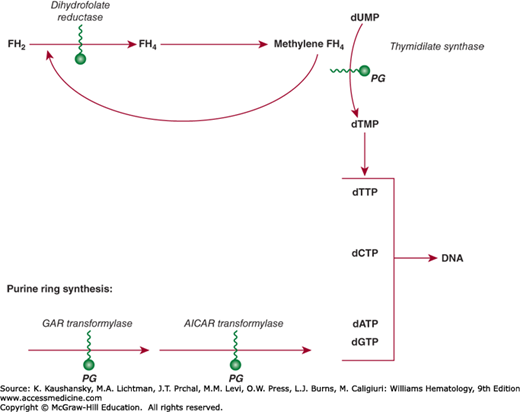
Methotrexate enters cells through an active uptake process mediated in most tumor cells by the reduced folate transporter20 and is actively effluxed from cells by the MRP class of exporters.21 A second uptake transporter, the membrane folate-binding protein (FBP), has lower affinity for methotrexate, but may contribute to uptake of other antifolates, such as pemetrexed. The FBP is found on many solid malignancies, and is an active target for folate analogue- and antibody-mediated drug development. A third, low pH transporter may also participate in methotrexate influx, particularly in the intestine, but its role in tumor uptake is uncertain.22 By virtue of its 4-amino substitution, methotrexate potently inhibits the enzyme DHFR, which recycles oxidized dihydrofolate to its active tetrahydrofolate state. Inhibition of DHFR leads to rapid depletion of the intracellular tetrahydrofolate coenzymes required for thymidylate and purine biosynthesis. As a result, DNA synthesis is blocked and cell replication stops. Methotrexate is retained intracellularly as a consequence of an enzymatic process that adds up to six glutamate moieties in an unusual peptide linkage to the γ-carboxyl group of the drug (Fig. 22–1). Polyglutamation is an important determinant of leukemic cell sensitivity to methotrexate. Methotrexate polyglutamates, in addition to their long persistence in cells and their potent inhibition of DHFR, have greatly increased inhibitory effects on other folate-dependent enzymes, including thymidylate synthase and enzymes that synthesize purines (Fig. 22–2). Cells that convert the drug to polyglutamates efficiently, such as leukemic myeloblasts and lymphoblasts, are more susceptible to the drug than are normal myeloid precursors, which have limited capability for polyglutamation.23 Accumulation of polyglutamates correlates with increased cytotoxicity and treatment response in childhood lymphoblastic leukemia.24 Hyperdiploid ALLs are particularly efficient in transporting methotrexate and in producing polyglutamated species, factors that may contribute to their favorable prognosis.25 Polyglutamates are slowly degraded to their readily effluxed monoglutamate form by γ-glutamyl hydrolase, and a polymorphism (T127I) that deceases γ-glutamyl hydrolase activity is associated with enhanced polyglutamate accumulation in leukemic cells.26 Acquired resistance to methotrexate in patients with leukemia is associated with several different alterations: increased levels of DHFR as a consequence of gene amplification,13 defective polyglutamation,27 impaired drug uptake,28 or increased efflux by the MRP class of transporters.29
Figure 22–1.
Structures of folate, tetrahydrofolate, and its analogue methotrexate. The vitamin is absorbed as a monoglutamate and converted intracellularly to a polyglutamate, in which form it is both physiologically active and is stored in cells. Methotrexate, the 2,4-diamino analogue of folic acid, is shown in the bottom panel and is also converted to a polyglutamate intracellularly. (Reproduced with permission from Brunton L, Chabner B, and Knollman B: Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 12th ed. New York, NY: McGraw-Hill; 2011.)
Methotrexate is well absorbed when administered orally at low doses (5 to 10 mg/m2), but when doses exceed 30 mg/m2, absorption is variable. Consequently, doses greater than 25 mg/m2 should be administered parenterally.
The concentration of methotrexate in plasma declines in a polyexponential manner. A very rapid initial disposition phase persists for only a few minutes after intravenous administration. The intermediate disposition phase has a 2- to 4-hour half-life and continues for 12 to 24 hours after dosing. The terminal phase of drug decay is considerably slower, with an 8- to 10-hour half-life, and this phase becomes important in determining drug toxicity and the effectiveness of leucovorin rescue in patients treated with high-dose methotrexate. Methotrexate is primarily excreted unchanged by the kidney, while a minor fraction of the drug (7 to 30 percent) is inactivated by hepatic hydroxylation at the 7 position. Thus, doses should be reduced in proportion to the decrease in creatinine clearance (CrCl) in patients with renal impairment (CrCl <60 mL/min), because the prolonged exposure to high blood levels may result in life-threatening hematologic and gastrointestinal toxicity.30 High-dose methotrexate (>0.5 g/m2) followed by leucovorin rescue is used to treat patients with high-grade lymphoma, osteosarcoma, and ALL.
In ALL, dose adjustment of methotrexate to maintain a specific area under the concentration × time (C × T) curve improves treatment outcome.31 Patients receiving high-dose methotrexate can be rescued from drug toxicity by administering small doses of N-10-formyltetrahydrofolate (leucovorin), which replenishes the intracellular pool of reduced folates. Leucovorin is administered intravenously or orally in doses of 10 to 15 mg/m2 at 6-hour intervals, starting 6 to 24 hours after the infusion of methotrexate, and continuing until plasma concentrations of the drug fall below 1 μM. In patients receiving high-dose methotrexate, drug levels are routinely assayed 24 to 48 hours after dosing to determine the rate of drug elimination and the safety of discontinuing leucovorin. Both methotrexate and its hydroxylated metabolite are organic acids, which, like uric acid, are much more soluble in alkaline urine. In patients receiving such therapy, renal toxicity may result from intrarenal precipitation of the parent drug or its 7-OH metabolite, and is generally the primary cause of decreased drug clearance and overwhelming toxicity. Renal dysfunction can be prevented by alkalinizing the urine to pH 7 with intravenous sodium bicarbonate prior to and during therapy. Patients should be given intensive hydration, as well. If drug concentrations in plasma exceed 1 μM at 48 hours after high-dose therapy, leucovorin should be continued at higher doses of 50 to 100 mg/m2 every 6 hours until methotrexate concentrations fall below 0.1 μM. The higher doses of leucovorin are necessary to compete with methotrexate for transport and polyglutamation. In cases of extreme renal failure, with stable drug levels in the 10 μM range, leucovorin will not be effective. In this setting, continuous flow hemodialysis may provide a sustained reduction in drug levels.32 An alternative effective measure in this circumstance is the administration of glucarpidase, a commercially available bacterial enzyme that instantly degrades antifolates33 and prevents further toxicity.
The dose-limiting toxicities of methotrexate are myelosuppression and gastrointestinal toxicity. Toxic doses of methotrexate can induce thrombocytopenia and/or leukopenia, although leukopenia is more common. An early indication of methotrexate toxicity to the gastrointestinal tract is oral mucositis, whereas more severe toxicity may be manifested as diarrhea and gastrointestinal bleeding. Less-common toxic effects of methotrexate are skin rash (10 percent), pneumonitis, and chemical hepatitis. Transaminase elevations are frequently seen after high-dose methotrexate but rapidly return to normal in most patients, and without sequelae, but low-dose chronic administration, as employed to treat psoriasis or rheumatoid arthritis, may lead to portal fibrosis and cirrhosis.
Methotrexate, given intrathecally in doses of 12 mg every 4 days for children older than age 3 years and for adults, is used to prevent or treat meningeal leukemia and lymphoma. Dose adjustment is required for children younger than age 3 years, and should be made according to established protocols. Because the drug distributes poorly into the ventricular system after spinal injection, patients with active meningeal leukemia are frequently treated through an indwelling ventricular reservoir. Toxicities caused by intrathecal administration of methotrexate include acute arachnoiditis with nuchal rigidity and headache, as well as more chronic CNS toxicities, such as dementia, motor deficits, seizures, and coma.34 Rarely, these neurotoxicities develop hours after intrathecal drug administration, but more commonly they occur in the days or weeks after initiation of intrathecal treatment, and are most often seen in patients with active meningeal leukemia. Leucovorin is ineffective in reversing or preventing these toxicities. Patients with such signs should undergo evaluation to rule out progressive CNS tumor, and if malignancy is not found, intrathecal cytarabine should be used for further therapy.
Methotrexate and 6-mercaptopurine (6-MP) are synergistic in their inhibition of purine biosynthesis. L-Asparaginase, an inhibitor of protein synthesis, blocks cells from entering DNA synthesis and antagonizes the effects of methotrexate, when used before the antifolate. The two drugs are not used concurrently.
Nonsteroidal antiinflammatory drugs, which diminish renal blood flow, may reduce methotrexate clearance, as may nephrotoxic antibiotics and platinum derivatives, and these or other renal toxins should be avoided in patients during high-dose methotrexate.
Ara-C is an antimetabolite analogue of cytidine, differing in the configuration at the substituent on C2′ position of the sugar, in which the C2′-hydroxyl group is cis-oriented relative to the C1′-N-glycosyl bond, in contrast to the trans configuration of the ribose nucleoside. Ara-C is a mainstay in the induction of remission in patients with acute myelogenous leukemia (AML).
High doses (1 to 3 g/m2) of intravenous ara-C given at 12-hour intervals for 6 to 12 doses are more effective alone or in a combination with anthracyclines than conventional doses (100 to 150 mg/m2 q12h) in consolidation therapy of AML, and they confer particular benefit in patients with cytogenetic abnormalities (t[8:21], inv[16], t[9:16], and del[16]) related to the core binding factor that regulates hematopoiesis.35 Other subsets of leukemia may have increased sensitivity to ara-C. ALL patients with mixed lineage leukemia (MLL) gene translocations have upregulation of the human equilibrative nucleoside transporter (hENT) and have a greater sensitivity to ara-C.36 AML patients with K-RAS gene mutations seem to derive greater benefit from high-dose ara-C than do patients with wild-type K-RAS in their tumors.37
Ara-C is converted to the nucleoside triphosphate, cytarabine triphosphate (ara-CTP) intracellularly. The first step is catalyzed by deoxycytidine kinase (dCK); polymorphisms of the dCK gene may affect the rate of activation, and ultimately response.38 Ara-CTP is an inhibitor of DNA polymerase and is also incorporated into DNA, where it terminates strand elongation.39 If repair is unsuccessful, apoptosis is initiated. Ara-C and its mononucleotide are deaminated and inactivated by two intracellular enzymes, cytidine deaminase and deoxycytidylate deaminase, respectively.
Acquired ara-C resistance in experimental leukemias consistently results from the loss of dCK.40 Other changes implicated in experimental tumors include decreased drug uptake because of decreased expression of the equilibrative nucleoside transporter, increased deamination, increased pool size of competitive deoxycytidine triphosphate, and inhibition of the apoptotic pathway. Some of these changes, particularly loss of dCK activity, have been reported in studies of human leukemia, but these results have not been confirmed in definitive trials.41
Ara-C is administered intravenously either as a bolus injection or, more commonly, as a continuous infusion. It is not orally bioavailable because of its degradation by cytidine deaminase, which is present in the gastrointestinal epithelium and liver. Two standard schedules of administration are used: (1) rapid infusion of 100 mg/m2 every 12 hours for 7 days; or (2) continuous infusion of 100 to 200 mg/m2 per day for up to 7 days. Ara-C distributes rapidly throughout total-body water and is eliminated from plasma with a biologic half-life of 7 to 20 minutes. Most of the dose is excreted as arabinosyluracil (ara-U), an inactive metabolite, which is formed in plasma, the liver, granulocytes, and other tissues. Inhibition of ara-C deamination by ara-U may be responsible for the prolongation of the biologic half-life of the drug as larger doses are administered.42 Single-bolus injections and short infusions (30 minutes to 1 hour duration) at doses as high as 5 g/m2 produce little myelotoxicity because of the drug’s rapid clearance, whereas continuous intravenous infusion of only 1 g/m2 over 48 hours produces severe marrow toxicity. High-dose ara-C (3 g/m2 q12h for 3 days on days 1, 3, and 5), is routinely used for consolidation therapy of AML, but lower doses of 1 gm/m2 or less should be used in patients older than 60 years to avoid CNS toxicity. Unlike most drugs, a relatively high concentration of ara-C is achieved in the cerebrospinal fluid after intravenous administration, and may approach 50 percent of the corresponding plasma concentration.
Ara-C is also used intrathecally to treat meningeal leukemia. Doses of 50 to 70 mg in adults are usually employed and afford cerebrospinal fluid levels of the drug near 1 mM, which decline with a half-life of 2 hours. Ara-C (50 mg given every 2 weeks) has been impregnated into a gel matrix, in a formulation called DepoCyt, for sustained release into the cerebrospinal fluid, thus avoiding the need for repeated spinal taps. Initial clinical results in spinal lymphomatous meningitis indicate that it has efficacy equal to that of methotrexate.43
The dose-limiting toxicity for conventional dosing regimens of intravenous ara-C, 100 to 150 mg/m2 per day for 5 to 10 days, is myelosuppression. Nausea and vomiting also occur at these doses, the severity of which increases markedly when higher doses are employed, although repeated administration of the drug results in some tolerance. The nadir of the white count and platelet count occurs at about days 7 to 10 after the last dose of drug. Cerebellar, gastrointestinal, and liver toxicity, as well as conjunctivitis have also been observed when high-dose regimens are used. Hepatotoxicity ranges from abnormalities in serum transaminase levels to frank jaundice. The severity of these effects increases as the duration of therapy is prolonged; however, toxic effects rapidly subside upon discontinuation of treatment. Pulmonary infiltrates as a result of noncardiogenic pulmonary edema, and occasionally associated with severe pulmonary dysfunction, occur in leukemic patients receiving ara-C, as do gastrointestinal ulcerations with bleeding and infrequently perforation. Ara-C treatment is also reported to predispose to Streptococcus viridans pneumonia.44
In patients older than 60 years of age, and in patients with renal dysfunction, intravenous high-dose ara-C (3 g/m2 every 12 hours, days 1, 3, and 5, for six doses) causes a high incidence of cerebellar toxicity, manifested as ataxia and slurred speech.45 Confusion and dementia may supervene, leading to a fatal outcome. Cerebellar toxicity is more frequent in patients with abnormal renal function because of slowed elimination of ara-U, with consequent inhibition of ara-C deamination. Intrathecal ara-C is usually well tolerated, but neurologic side effects have been reported (seizures, alterations in mental status).
Although primarily used for solid tumors, gemcitabine, a 2′-2′-difluoro analogue of deoxycytidine, has significant activity against Hodgkin lymphoma. Its mechanism of action is similar to ara-C, in that, as a triphosphate, it competes with deoxycytidine triphosphate for incorporation into the elongating DNA strand, where it terminates DNA synthesis. It is also self-potentiating in that at a second site of action, it inhibits ribonucleotide reductase and thereby reduces competitive pools of deoxycytidine triphosphate (dCTP). It achieves higher nucleotide levels in tumor cells than does ara-CTP, and has a longer intracellular half-life. Its clinical pharmacokinetics are determined primarily by its rapid deamination by cytidine deaminase, yielding a short plasma half-life (t1/2) of 15 to 30 minutes. Standard schedules use 1000 mg/m2 infused over 30 minutes, and produced peak drug concentrations of 20–60 μM in plasma. Longer infusion times may produce higher intracellular triphosphate concentrations, but the benefit is uncertain.46
Resistance in solid tumors arises from low expression of hENT, increased expression of ribonucleotide reductase, and low levels of the initial activating enzyme, dCK. Gemcitabine is an extremely potent radiosensitizer and should not be used concurrently with radiation therapy except in clinical trials.
Toxicities are acute myelosuppression, mild hepatic enzyme elevations, uncommonly a reversible pneumonitis, and with prolonged usage, a progressive hemolytic uremic syndrome with capillary leak, leading to pleural effusions, ascites, and renal failure.47
Both 5-azacytidine and decitabine (5-aza-2′-deoxycytidine), its closely related deoxy analogue, exhibit cytotoxic activity and also induce differentiation of malignant cells at low doses. The latter action results from their incorporation into DNA and their covalent inactivation of DNA methyltransferase. The resulting inhibition of methylation of cytosine bases in DNA leads to enhanced transcription of otherwise silent genes.48 The differentiating effects of 5-azacytidine are the basis for the induction of fetal hemoglobin synthesis in patients with sickle cell anemia and thalassemia53 and its approved use in low-dose therapy of myelodysplastic syndromes (MDS). The usual doses of 5-azacytidine are 75 mg/m2 subcutaneously or intravenously per day for 7 days, repeated every 28 days, whereas decitabine is used in doses of 20 mg intravenously every day for 5 days every 4 weeks. Responses become apparent in myelodysplasia after two to five courses.
5-Azacytidine and decitabine are rapidly deaminated to chemically unstable uridine metabolites that immediately degrade into inactive products. Pharmacologic activity results from phosphorylation of the parent compound by cytidine kinase (for 5-azacytidine) or dCK (for decitabine), with subsequent conversion to a triphosphate nucleotide that becomes incorporated into DNA. The primary clinical toxicities of both 5-azacytidine and decitabine49 include reversible myelosuppression, nausea and vomiting with higher doses, hepatic dysfunction, myalgia, and fever and rash. Resistance likely results from defects in drug activation or alternative mechanisms for gene silencing, such as histone methylation or acetylation.
Purine analogues (Fig. 22–3) occupy an important role in maintenance for childhood ALL, and in the past decade newer analogues have shown remarkable activity in chronic leukemias and small cell lymphomas. With methotrexate, 6-MP is a critical component in the maintenance phase of curative therapy of childhood ALL. Other purine analogues include azathioprine, a prodrug of 6-MP and potent immunosuppressive agent; allopurinol, an inhibitor of xanthine oxidase, useful in the prevention of uric acid nephropathy; 2-chlorodeoxyadenosine, effective in the treatment of hairy cell leukemia and other lymphoid malignancies; 6-thioguanine (6-TG), an infrequently used antileukemic agent; and fludarabine phosphate (2-fluoroara-adenosine monophosphate), an effective agent for chronic lymphocytic leukemia (CLL) and follicular lymphomas, and for suppression of graft-versus-host disease in transplantation. A new purine analogue, nelarabine, is an ara-guanine prodrug, with strong activity against T-cell diseases, including lymphoblastic leukemias and lymphomas.50 The basis for this T-cell sensitivity appears to be the resistance of arabinosylguanine (ara-G) to degradation by the catabolic enzyme, purine nucleoside phosphorylase. High levels of arabinosylguanine triphosphate (ara-GTP) accumulate in T-cell neoplasms, leading to Fas ligand-mediated apoptosis. The most recent addition, clofarabine, also an adenosine analogue, has notable activity against childhood ALL and adult AML. Deoxycoformycin, a potent inhibitor of adenosine deaminase, is also effective in the treatment of T-cell malignancies and hairy cell leukemia.
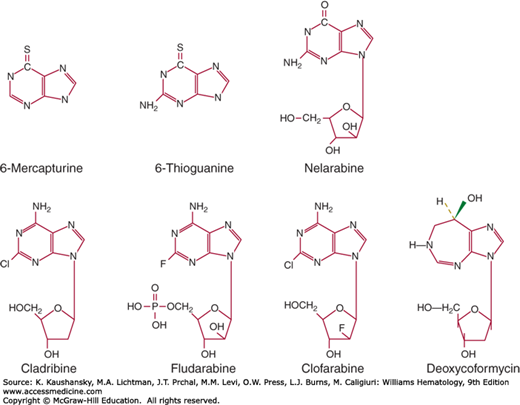
Both 6-MP and 6-TG have a thiol group substituted for the 6-hydroxy group of hypoxanthine or guanine, respectively, and are converted to nucleotides by hypoxanthine guanine phosphoribosyltransferase. They block synthesis of purines. The nucleotides of both 6-MP and 6-TG are incorporated into DNA, where they become methylated and are recognized by the mismatch repair system. Attempts to correct miscoding lead to strand breaks and apoptosis.51 Cell death correlates with the extent of their incorporation into DNA. 6-MP has the added effect of inhibiting de novo purine synthesis through the action of its metabolite, methyl-thioinosine monophosphate.52
In experimental tumor cells, resistance to 6-MP is most commonly caused by decreased activity of hypoxanthine guanine phosphoribosyltransferase (HGPRT), by increased efflux by the transporter MRP-4, and by the absence of an effective mismatch repair process. Resistance in human leukemia is poorly understood, but is linked to HGPRT deficiency. Patients differ in their rates of metabolic clearance of 6-MP and in their ability to efflux 6-thiopurines from cells. Rapid systemic clearance of the drug, as mediated by methylation of the thiol group by 5-thiopurine-methyltransferase (TPMT),53 is associated with a high leukemia recurrence rate in ALL maintenance therapy. Low levels of red blood cell thiopurine nucleotides correlate with a high level of activity of TPMT (more often found in patients of African descent) and a high risk of clinical relapse in patients with ALL,54 whereas decreased expression of TPMT, resulting from an inherited polymorphism in the number of tandem repeats in the 5′ promoter region, is associated with increased drug toxicity. A commercial test for enzyme polymorphism, based on red cell enzyme activity or thioguanine nucleotide content, is available. A second polymorphism of significance involves the cellular efflux protein, MRP-4; an inactive variant is associated with high 6-TG nucleotide concentrations in cells, and may be responsible for great sensitivity of Japanese patients to 6-thiopurines, as the variant occurs in 18 percent of the Japanese population.55 A polymorphism affecting the inosine triphosphate pyrophosphorylase enzyme (rs41320251) responsible for degrading a thiopurine nucleotide intermediate is associated with increased methyl-mercaptopurine nucleotides and a high incidence of febrile neutropenia in children with ALL.56
Methotrexate and 6-MP are highly synergistic, possibly because methotrexate blocks the de novo synthesis of purines, elevates phosphoribosyl pyrophosphate (PRPP), and enhances the activation of 6-MP. 6-MP blocks warfarin anticoagulation in some patients, leading to a requirement for higher doses of warfarin in patients receiving chronic 6-MP therapy for immunosuppression.
Both 6-TG and 6-MP are given orally at doses of 50 to 100 mg/m2 per day. Oral absorption of 6-MP is erratic, as only 16 to 50 percent of an oral dose is systemically available.57 Food and antibiotics may decrease absorption. Both 6-MP and 6-TG are inactivated by metabolism, and have half-lives of approximately 1 to 1.5 hour in plasma. Peak plasma levels of 6-MP occur 2 hours after administration and reach 1 to 2 μM. During 6-TG treatment, 6-TG nucleotides accumulate to much higher levels in leukemic cells, as compared to 6-MP. Inactive 6-thiomethyl nucleotides are almost 30-fold higher after 6-MP, than after 6-TG.58 6-MP is inactivated by metabolism to 6-thiouric acid, a reaction catalyzed by xanthine oxidase. Allopurinol inhibits the metabolic inactivation of 6-MP, but not of 6-TG. Therefore, it is generally recommended that dosages of orally administered 6-MP be reduced by 75 percent in patients receiving allopurinol. 6-TG is inactivated primarily by S-methylation, followed by oxidation and desulfuration, but a second pathway, mediated by guanase and xanthine oxidase, contributes to clearance. Dose reduction is not necessary when 6-TG and allopurinol are administered together.
Both 6-TG and 6-MP are myelotoxic, producing nadirs of white blood cells and platelets at 7 to 10 days after treatment.59 Moderate nausea and vomiting may also be observed. Patients may experience mild but rapidly reversible hepatotoxicity after treatment with either compound. Cirrhosis has occurred in some children with leukemia who are receiving long-term therapy with 6-MP. TPMT, which inactivates 6-thiopurines, occurs in several polymorphic forms that fail to metabolize the analogues. Approximately one person in 10 of the white population is heterozygous for ineffective polymorphic forms of the enzyme and will have significantly greater myelosuppression, whereas one patient in 300 is homozygous for the inactive forms, accumulates high concentrations of thioguanine nucleotides in both tumor and normal cells, and is at risk for overwhelming toxicity, even with greatly reduced doses of 6-MP.60
Other toxicities may include hypersensitivity reactions (fever, rash), interstitial pneumonitis; pancreatitis; opportunistic infection, and an increased incidence of AML in patients receiving chronic immunosuppressive treatment with 6-MP.
Originally synthesized as a deamination-resistant analogue of adenosine, fludarabine phosphate contains two important substitutions: a fluorine attached to the purine ring, which renders the drug resistant to deamination, and an arabinose sugar in place of deoxyribose, which leads to its pharmacologic activity as an inhibitor of DNA synthesis and ribonucleotide reductase. It has outstanding activity in CLL.61 It is strongly immunosuppressive, like the other purine analogues, and is frequently used for this purpose in nonmyeloablative allogeneic marrow transplantation62 and in the treatment of autoimmune diseases.
Activation of fludarabine phosphate requires removal of the phosphate group in plasma to allow cellular uptake by nucleoside transporters, and then intracellular rephosphorylation. Fludarabine is activated to the monophosphate level by dCK. The triphosphate inhibits DNA polymerase and becomes incorporated into both DNA and RNA.63 Its mechanism of cytotoxicity results from DNA chain termination and induction of apoptosis, although it also inhibits ribonucleotide reductase (RNR), a self-potentiating activity that decreases intracellular deoxyadenosine triphosphate (dATP) and increases fludarabine incorporation into DNA.64 Its triphosphate has a long intracellular half-life of 15 hours in CLL cells. Resistance has been ascribed to decreased active uptake, a deficiency of dCK, increased efflux, or increased RNR.
The drug is available in the United States as an intravenous preparation, and for oral use. It has 60 to 80 percent bioavailability. Because it is resistant to adenosine deaminase, fludarabine is eliminated primarily by renal excretion (60 percent), with a terminal half-life of 10 hours. For patients treated with fludarabine, the standard intravenous dose is 25 mg/m2 daily for 5 days, whereas the approved oral dose is 40 mg/m2 daily for 5 days. In patients with renal impairment, a 20 percent dose reduction for a CrCl of 17 to 40 mL/min/m2, and a 40 percent dose reduction for a CrCl less than 17 mL/min/m2 yields an area under the curve approximately equal to that seen in patients with normal renal function receiving full doses of fludarabine.65,66
When administered at these doses, fludarabine causes only moderate myelosuppression. In CLL patients, its antileukemic effect will lead to a progressive improvement in marrow function over a period of two to three cycles of treatment, with a median time to disease progression of 31 months. However, the drug also exerts cytotoxic effects against both B and T lymphocytes, lowering CD4 T-cell counts to 150 to 200 cells/μL and predisposing patients to opportunistic infections. In patients with a large tumor burden, rapid tumor lysis may rarely lead to hyperuricemia, renal failure, and hypocalcemia (tumor lysis syndrome).67 Thus, patients should be well hydrated and their urine alkalinized prior to beginning therapy. The primary acute toxicity is reversible myelosuppression. Peripheral sensory and motor neuropathy may occur during standard-dose therapy; autoimmune phenomena, including prolonged hypothyroidism, neutropenia and hemolytic anemia with both warm and cold antibodies, have been reported.68 Approximately 10 percent of CLL patients receiving fludarabine may develop a hypersensitivity syndrome of pulmonary infiltrates, hypoxemia, and fever, responsive to glucocorticoids.69 Myelodysplasia and acute leukemias, with chromosome 7p deletions, have been reported as infrequent late complications.70
The extreme sensitivity of normal and malignant lymphocytes to deamination-resistant purine analogues is further exemplified by the potent activity of cladribine in hairy cell leukemia, CLL, and low-grade lymphomas.71 A single course of cladribine, typically 0.09 mg/kg per day for 7 days by continuous intravenous infusion, induces complete response in 80 percent of patients with hairy cell leukemia. Administration by subcutaneous injection or by 2-hour intravenous infusion daily for 5 days to the same total dose achieves similar results. The drug has much the same intracellular fate as fludarabine, undergoing phosphorylation by dCK and further conversion to a triphosphate that becomes incorporated into DNA. The triphosphate of cladribine has a long intracellular half-life of 9.7 hours in CLL cells isolated from patients treated with the drug.72 The triphosphate has multiple metabolic effects, disrupting oxidative phosphorylation in mitochondria, inhibiting RNR and depleting nicotinamide adenine dinucleotide levels in tumor cells. All of these actions may explain the drug’s toxicity to slowly dividing lymphoid malignancies such as hairy cell leukemia and CLL. The actual mechanisms by which cladribine induces DNA strand breaks are not completely understood. However, similar to fludarabine, it inhibits DNA chain extension and daughter strand synthesis.73 Furthermore, the drug’s inhibition of RNR lowers levels of the competitive dATP. The cumulative effects of cladribine induce apoptosis (programmed cell death).
Cladribine is eliminated primarily (>50 percent) by renal excretion, with a terminal plasma half-life of 7 hours. In a patient with renal failure, continuous flow hemodialysis effectively cleared the drug and prevented serious myelosuppression.74 Cladribine retains effectiveness in at least a fraction of hairy cell leukemia patients resistant to deoxycoformycin or fludarabine, although clinical experience with sequential use of these drugs is limited. Toxicities of cladribine include transient myelosuppression, fever, tumor lysis syndrome, and occasional opportunistic infections possibly related to immunosuppression. The development of cumulative thrombocytopenia during treatment with repeated courses of the drug may limit its use. Resistance develops in experimental tumors through decreased uptake, loss of the activating enzyme dCK, increased RNR activity, increased efflux,75 or by induction of 5′-nucleotidase activity.
This analogue has halogen substitutions on both the purine ring and arabinose sugar, resulting in a ready uptake and activation, to a highly stable intracellular triphosphate (half-life of 24 hours), which terminates DNA synthesis, inhibits RNR, and induces apoptosis. The usual adult dose is 52 mg/m2 given as a 2-hour infusion daily for 5 days. Clofarabine has a plasma half-life of 6.5 hours. The primary route of clearance is through renal excretion, and dose adjustment according to CrCl is recommended for patients with abnormal renal function.
Toxicities are myelosuppression; uncommonly, fever, hypotension, and pulmonary edema, suggestive of capillary leak caused by cytokine release; hepatic transaminitis; hypokalemia; and hypophosphatemia. As a single agent, the drug is well tolerated as second-line treatment for AML patients with remission rates of 30 percent.76
A guanine nucleoside analogue, nelarabine has useful activity as a secondary agent for T-cell lymphoblastic lymphoma and acute T-cell leukemias. Its mode of action is similar to the other purine analogues, in that it becomes incorporated into DNA and terminates DNA synthesis. Its selective action for T cells may relate to the ability of T cells to activate purine nucleosides and the lack of susceptibility of this drug to purine nucleoside phosphorylase, a degradative reaction.
Usual doses are an intravenous 2-hour infusion of 1500 mg/m2 for adults on days 1, 3, and 5, and a lower dose of 650 mg/m2 per day for 5 days for children. The drug is rapidly demethylated by adenosine deaminase after administration, yielding the ara-G, which is cleared by hydrolysis and has a longer plasma half-life of 3 hours. ara-G is converted intracellularly to its triphosphate77 which becomes incorporated into DNA. The primary toxicities are myelosuppression and abnormal liver function tests, but the drug may cause a spectrum of neurologic abnormalities, including seizures, delirium, somnolence, and the Guillain-Barré syndrome of ascending paralysis.
Pentostatin contains a unique seven-carbon primary ring system that closely resembles the transition-state intermediate of the adenosine deaminase reaction. As such, pentostatin is a potent inhibitor of the enzyme, leading to accumulation of intracellular adenosine and deoxyadenosine nucleotides. In addition, the triphosphate of pentostatin is incorporated into DNA. The imbalance in purine nucleotide pools produced by pentostatin probably accounts for its cytotoxicity.
Although initial trials of pentostatin demonstrated striking renal and neurologic toxicities at doses of 10 mg/m2 intravenously per day or greater, lower doses (4 mg/m2 biweekly) are extremely effective in inducing pathologically confirmed complete responses in hairy cell leukemia. At this lower dose, severe depletion of normal T cells occurs and may predispose to opportunistic infection.78 The optimal dose may be lower than 4 mg/m2 biweekly. The drug is eliminated entirely by renal excretion, necessitating proportional dose reduction in patients with reduced CrCl.
Hydroxyurea inhibits RNR, the enzyme that converts ribonucleotide diphosphates to deoxyribonucleotides. It chelates iron, an essential cofactor in the RNR reaction. In malignant disease, hydroxyurea is most commonly used for treating polycythemia vera, essential thrombocythemia, and the chronic phase of CML and to lower the myeloblast count in patients presenting with AML or blastic crisis of CML. It has also become the standard agent for preventing painful crisis and reducing hospitalization in patients with sickle cell disease and in thalassemia patients with hemoglobin (Hgb) C/SS. Its antisickling activity results from induction of Hgb F through its activation of a specific promoter for the γ-globin gene. It may also exert antisickling activity and decrease occlusion of small vessels through its generation of nitric oxide, a vasodilator, and through decreased expression of adhesion molecules such as L-selectin, on neutrophils.79 Resistance occurs in experimental tumors as a consequence of amplification of the catalytic subunit of RNR or through mutations in RNR that lower affinity for the enzyme.
Hydroxyurea is well absorbed orally, even when large doses such as 50 to 75 mg/kg orally are given for rapid lowering of the white blood cell count. In chronic therapy of myeloproliferative neoplasm, starting doses of 15 mg/kg orally are adjusted upward or downward based on neutrophil counts. In managing patients with sickle cell disease, neutrophils should be maintained above 2000 per mL.80 Hydroxyurea may also be given intravenously to rapidly lower the white blood cell count in patients with extreme leukemic leukocytosis or thrombocytosis. Peak plasma levels following oral administration are achieved at about 1 hour and decline with a half-life of 3 to 4 hours thereafter. Renal excretion is the major route of drug elimination, and doses should be decreased in proportion to the deficit in CrCl.
The major toxicities of hydroxyurea are leukopenia and the induction of megaloblastic changes. Nausea, drug fever, pneumonitis, maculopapular skin rash, and painful leg ulcers have been observed with this drug, although it is generally well tolerated. Hydroxyurea, like ara-C, is an S-phase–specific agent. Accordingly, single large doses cause little toxicity other than myelosuppression. The nadir of the leukocyte count occurs 3 to 5 days after a single dose of drug, and the leukocyte count recovers rapidly. It is a potent teratogen and should not be used in women of childbearing age. Its potential to cause leukemic transformation is uncertain, but small cases series suggest this may occur in patients with a myeloproliferative neoplasm.81
ANTITUBULINS
Vinblastine and vincristine are commonly used in the treatment of hematologic neoplasms: vinblastine because of its excellent activity in the treatment of Hodgkin lymphoma and vincristine in lymphomas and childhood leukemia. Both drugs have activity in solid-tumor therapy, particularly in treating childhood sarcomas (vincristine), and testicular cancer (vinblastine).
The vinca alkaloids exert their cytotoxic action by their binding to tubulin, a structural protein found in the cytoplasm of cells. Microtubules, assembled through polymerization of tubulin dimers, form the spindle along which the chromosomes migrate during mitosis. Microtubules are an important structural component of neuronal axons. Binding of the vinca alkaloids to tubulin leads to inhibition of formation of the mitotic spindle,82 arresting cells in metaphase and inducing apoptosis. Resistance to the vinca alkaloids may be acquired through the expression of the MDR efflux pump. Alternatively, resistant cells may contain mutant tubulin with decreased avidity of vinca binding.83 The clinical importance of these resistance mechanisms, however, is uncertain.
Vincristine and vinblastine are both administered by the intravenous route. The average single dose of vincristine is 1.4 mg/m2 and that of vinblastine 8 to 9 mg/m2. Sequential doses of the drugs are usually given at 1- or 2-week intervals. These doses provide peak plasma drug concentrations of approximately 1 μM. The plasma pharmacokinetics of both vinca analogues are characterized by a very rapid initial disposition phase followed by a slow terminal phase of decay, with half-lives of 20 to 85 hours. Almost 70 percent of a dose of vincristine is metabolized by the liver and excreted in the feces. Cytochrome P450 (CYP)-mediated metabolism is also the major route of inactivation of vinblastine, producing a variety of inactive metabolic produced in the liver are excreted in the bile. Inducers of CYP3A4, such as phenylhydantoin, enhance clearance, while inhibitors delay clearance and increase toxicity. The dose of vincristine or vinblastine should be reduced in patients with hepatic impairment. Although specific guidelines for dose reduction have not been developed, a 50 percent decrease in dose is recommended for patients presenting with a bilirubin level of 1.5 to 3 mg/dL and a 75 percent reduction for levels greater than 3 mg/dL. Dose reduction is not necessary for patients with impaired renal function, as very little intact drug is excreted in urine.
The dose-limiting side effect of vincristine is neurotoxicity, which usually occurs when the total dose received exceeds 6 mg/m2. The initial signs of neurotoxicity are paresthesia of the fingers and lower extremities and loss of deep tendon reflexes. Continued administration may lead to profound loss of motor strength, such as weakness of dorsiflexion of the foot and extension of the wrists. Elderly patients are particularly susceptible to such toxicities. Occasionally, cranial nerve palsies may lead to vocal cord paralysis or diplopia, and severe jaw pain may result from vincristine administration. At high doses of vincristine (>3 mg total single dose), autonomic neuropathy may cause obstipation and paralytic ileus. Sensory changes and reflex abnormalities slowly improve when the drug is discontinued; motor impairment improves less rapidly and may be irreversible. Inappropriate antidiuretic hormone release resulting in symptomatic dilutional hyponatremia has been ascribed to vincristine.
While marrow suppression is not common with vincristine administration, myelosuppression may be noted in patients with impaired marrow function as a consequence of prior treatment with other drugs. Platelet counts are relatively unaffected.
The primary toxicity of vinblastine is leukopenia. The white count reaches a nadir at day 7 and reverses rapidly thereafter. Mucositis may result from higher doses (>8 mg/m2) of vinblastine or when it is used in combination with other cytotoxic drugs. Neurotoxicity is rare, but ileus can occur at high doses.
Both drugs cause severe pain and local toxicity if extravasated. Neither drug should ever be given intrathecally. Vincristine administered inadvertently into the cerebrospinal fluid causes acute neurologic dysfunction, coma, and death. Attempts at replacement of the cerebral spinal fluid with an electrolyte solution, Ringer lactate, supplemented with 15 ml/L of fresh-frozen plasma, have been reported to avert a fatal outcome, but do not prevent severe neurologic sequelae.84
The taxanes, paclitaxel, docetaxel, and Abraxane, are a second class of antimitotic compounds that differ in mechanism and toxicity profile from the vinca alkaloids that are primarily used in patients with solid tumors. Paclitaxel was purified from an extract of the bark of Taxus brevifolia, whereas docetaxel is a closely related semisynthetic derivative. Abraxane is paclitaxel embedded in an albumin microparticle. The taxanes bind to the β-tubulin subunit of microtubules and promote the polymerization of microtubules, leading to disordered mitotic spindle formation and a block in the progression through mitosis.85 They induce apoptosis in tumor cells irrespective of the p53 status of the cells and kill cells at 10 nM concentrations or less in cell culture in a time-dependent manner.86 In experimental settings, resistance is related to increased drug efflux, mutations in β-tubulin, or increased expression of antiapoptotic proteins such as survivin,87 or of the mitosis-related aurora kinase.88
The taxanes are subject to MDR mediated by the mdr and mrp genes, as well as to β-tubulin mutations. Because they are highly insoluble in aqueous solution, paclitaxel and docetaxel are formulated in lipid-based solvents that cause occasional hypersensitivity reactions. Thus, paclitaxel is given after pretreatment with antihistamines (cimetidine, diphenhydramine), and dexamethasone. Both drugs are cleared primarily by hepatic CYP metabolism, although by different isoenzymes (paclitaxel predominantly by CYP2B6 and docetaxel by CYP3A4) with terminal plasma half-lives of 10 to 13 hours. Their metabolism is stimulated by phenytoin and other CYP-inducing drugs and inhibited by ketoconazole. Their major toxicities, aside from hypersensitivity, are a sharp but brief leukopenia, milder thrombocytopenia, and mucositis. High-dose or repeated cycles of the taxanes cause a sensory and motor peripheral neuropathy that is reversible with drug discontinuation. Occasional patients have experienced atrial conduction block or atrial or ventricular arrhythmias after paclitaxel administration, and the combination of paclitaxel with doxorubicin may produce a greater incidence of congestive heart failure than seen with doxorubicin alone.89 A syndrome of progressive fluid retention and peripheral edema occurs in patients receiving multiple cycles of docetaxel and can be at least partially prevented by pretreatment with glucocorticoids.90
The taxanes have not found a valuable role in the treatment of hematologic malignancy. However, a number of analogues and new formulations are under development. Abraxane, consisting of paclitaxel bound to albumen microparticles, does not require a lipid solvent, is virtually free of hypersensitivity as a side effect, and enters cells by a separate albumen-mediated transporter. It is approved for treatment of relapsed breast cancer and pancreatic cancer. Cabazitaxel, approved for prostate cancer, is a new analogue with decreased susceptibility to MDR. An entirely new class of natural products, the epothilones, have a similar mechanism of action, are less susceptible to MDR, and have activity against breast cancer.91
TOPOISOMERASE INHIBITORS
This group of compounds includes synthetic derivatives of 20 (S)-camptothecin, a naturally product from the Camptotheca acuminata tree. The camptothecins interact with a unique target, topoisomerase I, stabilizing the enzyme’s complex with DNA and preventing the resealing of DNA single-strand breaks induced by the enzyme. Resistance arises through mutation, deletion, or decreased expression of the topoisomerase I gene. The primary agents in clinical use are irinotecan, which is approved for treatment of colon cancer, and topotecan, approved for second-line treatment of ovarian cancer and small cell lung cancer. Irinotecan, most commonly administered intravenously at a dose of 125 mg/m2 once each week for 4 weeks every 42 days, has shown promise against lymphomas in phase II trials performed in Japan.92 Response rates of 42 percent in previously treated patients with non-Hodgkin lymphoma, and of 38 percent in patients with refractory or relapsed adult T-cell leukemia-lymphoma, remain to be confirmed. Topotecan has remission-inducing activity in patients with myelodysplasia and chronic myelomonocytic leukemia, both as a single agent (1.5 mg/m2 per day for 5 days) and in combination with ara-C.93,94 Objective responses have also been observed in phase I clinical trials in patients with AML.95 Irinotecan and topotecan differ substantially in their profile of toxicities and pharmacokinetic behavior. Irinotecan is a water-soluble prodrug that is converted to the active species, SN-38, by carboxyl esterase-mediated cleavage. Irinotecan and SN-38 are both eliminated by glucuronidation and biliary excretion. Therefore, irinotecan must be used with caution and at lower doses in patients with Gilbert disease (and lacking glucuronyl transferase 1A1) or in those with hepatic dysfunction.96 In contrast to the hepatic extraction and excretion of irinotecan, approximately two-thirds of the dose of topotecan is eliminated by renal excretion, with the remainder being cleared by biliary excretion. Dose adjustment proportional to CrCl is indicated in patients with renal failure.97 Topotecan toxicity consists mainly of myelosuppression and, to a lesser degree, mucositis, whereas irinotecan causes a profound diarrhea, which is responsive to loperamide, and a more modest myelosuppression. The maximum tolerated dose of topotecan for the 5-day schedule of 30-minute intravenous infusions/day is 4.5 mg/m2 per day in patients with leukemia.98 This is considerably greater than the approved dose for solid tumors, and gastrointestinal side effects, such as mucositis and diarrhea, become dose-limiting at these higher doses.
The anthracyclines are a unique class of natural products that inhibit topoisomerase II (Topo II), an enzyme important in DNA strand passage allowing the untangling of DNA prior to replication or repair. Doxorubicin, daunorubicin, idarubicin, and epirubicin are closely related in structure, each possessing a rigid planar core to which is linked a daunosamine sugar. The molecules differ in side-chain substitutions attached to the anthracycline ring system, and exhibit different spectra of antitumor activity and toxicity. Mitoxantrone, a closely related, nonglycosidic anthracenedione, has very similar pharmacologic properties to those of the anthracyclines. The anthracyclines are produced by a Streptomyces species, whereas mitoxantrone is a synthetic compound. Doxorubicin (Adriamycin) has broad activity against solid and hematologic malignancies. It is an important component of the standard multidrug regimens used to treat Hodgkin lymphoma (doxorubicin, bleomycin, vinblastine, and dacarbazine [ABVD]) and aggressive non-Hodgkin lymphoma (cyclophosphamide, doxorubicin, vincristine, and prednisone). Daunorubicin and idarubicin are used almost exclusively in combination with ara-C for the treatment of AML, whereas epirubicin is primarily effective against solid tumors. Mitoxantrone is employed for the treatment of AML and breast cancer, and as an immunosuppressive for patients with multiple sclerosis. Liposome-encapsulated doxorubicin (Doxil) and daunorubicin derivatives are approved for treatment of solid tumors; they provide a more prolonged, lower peak concentration of drug, and have decreased cardiac toxicity. The daunorubicin liposome is of interest in treating AML.99 A novel anthracycline, pixantrone, which has lesser cardiotoxicity, has received conditional approval in Europe for refractory non-Hodgkin B-cell lymphoma.100
Anthracyclines target the replication and structural integrity of DNA. Their primary mechanism of toxicity stems from their interaction with Topo II, an enzyme that creates DNA strand breaks and promotes strand passage through those breaks. Strand passage is essential in untangling DNA in preparation for replication and repair. Once the strand passage and unwinding is complete, Topo II reseals the broken DNA strands. The anthracyclines inhibit the resealing step by forming a complex with Topo II and the broken DNA strand to which the enzyme is linked. Accumulation of strand breaks activates apoptosis. The planar molecular structure of these drugs promotes their intercalation between opposing strands of the DNA helix and may contribute to the specificity of sites of DNA breakage. In addition to their inhibition of Topo II, anthracyclines generate free radicals by virtue of the oxidation-reduction cycling of their quinone group, an action catalyzed by the binding of Fe2+. Free radical generation is thought to be responsible for their cardiac toxicity.
The importance of the presence of Topo II in determining response to anthracyclines is best illustrated by the greater benefit of anthracycline-based breast cancer treatment in patients with amplification of the target enzyme on chromosome 17, near the HER2 gene with which it coamplifies.101 Anthracycline-containing regimens are particularly effective in HER2-amplified breast cancers.102
Anthracyclines enter cells through a passive transport process. Their lipophilic structure allows them to achieve high intracellular concentrations. Anthracyclines are pumped out of the cell by a series of ATP-dependent transporters, including the P-glycoprotein MDR transporter, the breast cancer resistance protein transporter (BCRP) and related efflux pumps.11 Other mechanisms for anthracycline resistance include decreased Topo II activity or Topo II mutations in the enzyme that inhibit drug binding, as well as defects in apoptosis or impaired checkpoint recognition of DNA strand breaks.
Daunorubicin and idarubicin are readily converted to active hydroxyl metabolites, whereas doxorubicin produces limited amounts of alcohol metabolite. The alcohols of daunorubicin and doxorubicin are less active as antitumor agents, but do possess cardiotoxic activity.
All anthracyclines are eliminated by the formation of inactive metabolic products (aglycons, side-chain-modified products, glucuronides, sulphates, and oxidative metabolite) in the liver. Only a minor fraction of the dose of any of the anthracyclines is excreted in the urine as the parent drug or alcohol metabolite. The pharmacokinetics of the clinically useful anthracyclines are predominantly influenced by their terminal disposition phase, which exceeds 24 hours. Although prolongation of the half-life of doxorubicin has been reported in studies of patients with compromised liver function, no clear correlations of liver function with toxicity have been established. However, in patients with elevated serum bilirubin levels, initial doses of doxorubicin and daunorubicin should be reduced by 50 percent, and adjust there after according to tolerance. Idarubicin, the only anthracycline amenable to oral administration, has a bioavailability of 20 percent for the parent drug and 40 percent for parent drug plus idarubicinol, the primary active metabolite. Idarubicinol has a very prolonged biologic half-life, ranging from 50 to 60 hours, and is likely responsible for the antitumor activity of this drug. In contrast to the metabolites of doxorubicin and daunorubicin, idarubicinol is eliminated primarily by renal excretion. No dose adjustment for hepatic dysfunction is indicated.
Mitoxantrone has a long terminal half-life of 23 to 42 hours. Only a minor fraction of unchanged drug is excreted in the urine (<10 percent) or stool (<20 percent). The majority of the drug is metabolized or bound to tissues. Patients with impaired hepatic function may have a more prolonged elimination of mitoxantrone.
The usual dose of doxorubicin when administered as a single agent by bolus intravenous injection is 45 to 75 mg/m2 every 3 to 4 weeks, depending on the tumor treated and the drug combination. Less cardiac toxicity may result from schedules that avoid high peak plasma concentrations, such as weekly doses (15 to 25 mg/m2) or continuous intravenous infusion over 48 to 96 hours, as in the EPOCH (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) regimen.103 When given in combination with other myelotoxic agents such as cyclophosphamide, the dose of doxorubicin is usually decreased because of overlapping marrow toxicity. Daunorubicin has been used as the anthracycline of choice in the treatment of AML. To minimize the cardiotoxic effects of daunorubicin, in standard “3+7” therapy in AML the daunorubicin dosage (45 to 60 mg/m2 for adults <60 years) is given daily for 3 days to avoid high peak concentrations, although larger doses (90 mg/m2/day), may produce higher complete remission rates. In the elderly, a lower dose of 30 mg/m2 daily for 3 days is typically used in combination therapy.
Myelosuppression is the primary acute toxicity of this class of drugs, with a nadir occurring 7 to 10 days after single-dose administration and recovery by 2 weeks. Mitoxantrone produces less nausea and vomiting than does either daunorubicin or doxorubicin. Doxorubicin may cause mucositis, especially when used in maximally tolerated divided doses given over 2 to 3 days or when used in combination with other drugs that cause mucositis. Anthracyclines can also cause radiation recall in previously irradiated tissues, especially when the drug is administered just prior to or in the weeks following irradiation. Alopecia often occurs. Extravasation of these drugs can result in tissue necrosis so they should be administered through an indwelling central venous catheter. Dexrazoxane injected subcutaneously, lessens tissue damage after extravasation.104 Patients receiving doxorubicin should also be warned that their urine may turn red.
Cardiotoxicity is the major late toxic effect of anthracyclines.105 Cardiotoxicity most likely results from free radical formation catalyzed by the anthracycline’s quinone moiety, although cardiac Topo IIb (not the major topoisomerase involved in DNA replication) may play a role in mediating this effect. Iron as a reduction-oxidation (redox) cofactor contributes to toxicity, as it accumulates in mitochondria during treatment.106 Cardiac proteins, including the cardiac myosin-binding protein C, show evidence of alkylation and degradation after anthracycline treatment.107
Clinically, anthracycline-induced cardiotoxicity presents after repeated cycles of treatment. Rarely, acute effects are manifest as arrhythmias, conduction abnormalities, or a “pericarditis–myocarditis syndrome.” The more common long-term consequence is congestive heart failure (CHF), which can develop during or several months after treatment. Studies in breast cancer have demonstrated a 0.5 to 1 percent risk of cardiomyopathy in patients treated with adjuvant anthracyclines.108 The risk is higher in patients receiving trastuzumab or paclitaxel in combination with doxorubicin.
The risk of anthracycline-induced cardiotoxicity increases with total dose, but is difficult to estimate for any individual patient. In patients with normal cardiac function prior to treatment, the subsequent rate of doxorubicin-induced CHF reaches less than 1 percent at total doses of 400 mg/m2, but climbs steeply thereafter to 7 to 20 percent at total doses of 550 mg/m2.109 The threshold for cardiotoxicity varies among the different anthracyclines. For example, the inflection threshold for daunorubicin (600 to 700 mg/m2) is significantly higher than for doxorubicin (400 mg/m2). However, it should be remembered that these thresholds are based on population studies, and for any individual patient the risk is impossible to predict. The clinician must pay close attention to symptoms of CHF, such as dyspnea, cough, orthopnea, and weight gain or ankle edema, throughout a course of treatment and irrespective of total dose.
Besides the cumulative dose of anthracycline, other risk factors for anthracycline-induced cardiomyopathy include mediastinal (mantle) radiation, preexisting heart disease, and patient age, the risk being highest in children younger than the age of 4 years. Children who receive greater than 300 mg/m2 have a significant risk of having decreased myocardial contractility, decreased ventricular dimension, and an increased incidence of cardiac events (such as conduction defects, myocardial infarction, and CHF) in their adult years. The incidence of may be decreased by coadministering dexrazoxane, an iron chelator, during chemotherapy.110 It is recommended that the total doxorubicin dose be limited to 300 mg/m2 in children. In addition, children treated with anthracyclines should have long-term cardiology followup.110
Ejection fraction measurements have been helpful in detecting a decline in myocardial function, a sign of impending myocardial failure. Ejection fraction measurements, usually by multigated acquisition scan (MUGA), should be performed to verify normal cardiac function prior to starting anthracycline-based chemotherapy, and should be repeated at the earliest clinical sign of cardiac dysfunction, and before every two cycles of treatment when the total dose of doxorubicin exceeds 300 mg/m2. Anthracyclines should be discontinued if the ejection fraction falls below 40 percent, or if the ejection fraction drops a total of 20 percent from pretreatment levels.
As cardiotoxicity of anthracyclines results from the generation of free radicals by an anthracycline–iron complex, dexrazoxane, an iron chelator, decreases free radical formation in vitro and decreases the risk of cardiotoxicity in children receiving treatment for ALL, and in adults with metastatic breast cancer.110 Fortunately, dexrazoxane does not cause any apparent diminution of antitumor activity. Adding dexrazoxane to an anthracycline-based regimen represents an alternative to discontinuing anthracyclines in patients who are approaching total-dose thresholds of drug, but who still require treatment. Two trials have shown a higher rate of secondary leukemia and MDS in patients receiving doxorubicin and dexrazoxane.111,112 In adult patients, dexrazoxane should be added only in patients who have received a total dose of at least 300 mg/m2 doxorubicin or 540 mg/m2 epirubicin.
Treatment with Topo II inhibitors, including anthracyclines, mitoxantrone, and the epipodophyllotoxins (see “Epipodophyllotoxins” below), increases the risk of AML. AML typically develops 6 months to 5 years after exposure to the Topo II inhibitor.107 This heightened risk derives from the increased DNA double-strand breaks generated by the Topo II inhibitor. These double-stranded DNA breaks can give rise to balanced chromosomal translocations involving the MLL or PML genes.111 Anthracyclines and mitoxantrone have affinity for specific DNA sequences and cause translocations at specific hot spots in the genome, including a 6-base pair breakpoint region in the PML gene causing the 15;17 translocation, the 11q23 translocation involving the MLL gene, and the 11;20 translocation involving the NUP98 gene.113,114
Two semisynthetic derivatives of podophyllotoxin, VP-16 (etoposide) and VM-26 (teniposide), inhibit Topo II and have significant clinical activity in hematologic malignancies. Etoposide has been incorporated into combination therapy regimens for Hodgkin lymphoma, large cell non-Hodgkin lymphomas, leukemias, and various solid tumors, and is a frequent component of high-dose chemotherapy regimens. Teniposide has limited value in clinical oncology. Its use is generally restricted to childhood acute leukemia, where it appears to be synergistic with ara-C. These compounds induce double-stranded breaks in DNA through their sequence-specific binding to DNA in complex with Topo II.115 One mechanism of resistance is increased expression of the MDR drug exporter.11 A second mechanism results from decreased Topo II activity or mutation of the enzyme, resulting in decreased drug binding.116,117
Etoposide is administered in doses of 100 to 120 mg/m2 per day for 3 days, either consecutively or every other day. Approximately 30 to 40 percent of an intravenous dose of etoposide is excreted intact in the urine, while the remainder is cleared by hepatic glucuronidation or demethylation; thus, doses of etoposide require modification for patients with compromised renal or hepatic function.118 The plasma half-life of etoposide is 15 hours. The clinical activity of etoposide is highly schedule dependent. Single conventional doses are essentially without antitumor effect as compared to consecutive daily doses for 3 to 5 days. The pharmacokinetics of teniposide are very similar to those of etoposide, with a terminal plasma half-life of 20 to 48 hours. However, little parent drug appears intact in the urine, and dose modification for patients with renal dysfunction is unnecessary.
When administered intravenously, both etoposide and teniposide should be infused over a 30-minute period to avoid hypotensive episodes. The major toxicity of both drugs is leukopenia, which is rapidly reversible; thrombocytopenia is less common. Nausea and vomiting often follow etoposide administration. Alopecia may occur with both drugs. Other toxicities, such as fever, mild elevation of liver function tests, and peripheral neuropathy, are relatively uncommon. Because the major toxicity of etoposide is limited to the marrow, this drug is a valuable component of high-dose regimens used with marrow transplantation. In high-dose etoposide protocols (1.5 g/m2 or greater given over 3 to 5 days) oropharyngeal mucositis becomes a prominent toxicity. Less-frequent high-dose toxicities include hepatocellular damage and, rarely, anaphylactic-like symptoms, probably related to the formulation vehicle. Secondary AML associated with translocation at 11q23 or the PML gene may follow etoposide treatment in children with ALL119 and in adults with solid tumors.120
AGENTS ACTIVE THROUGHOUTTHE CELL CYCLE
These drugs are important in the treatment of hematopoietic malignancies either as single agents or as components of standard- or high-dose regimens. Their role as treatment for both acute and chronic hematologic malignancies results from their unique mechanism of cell killing and their lack of cell-cycle specificity. They may eradicate noncycling cells that escape cycle-active components of the treatment. Although these agents share the common property of forming covalent bonds with electron-rich sites on DNA (oxygen and nitrogen substituents), they exhibit important differences in their intrinsic reactivity, route of cellular uptake, favored sites of alkylation on DNA bases, and the specific mechanism of DNA repair that determines cell survival. These differences are borne out in experimental settings, where cross-resistance to alkylating agents is incomplete. Thus, protocols employing multiple alkylators, particularly in high-dose regimens, have a rational basis.121
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


