PATHOGENESIS AND PATHOPHYSIOLOGY OF BACTERIAL INFECTIONS
PHILIPP AGYEMAN, DENIS GRANDGIRARD, AND STEPHEN L. LEIB
The brain not only is a normally sterile site but also is protected from infection by specialized barriers, including the bony skull and the WWWblood–brain barrier (BBB). Consequently, infections of the central nervous system (CNS) in general and bacterial infections of the CNS in particular are comparatively rare. In the United States, the incidence of bacterial meningitis decreased to 1.38 per 100,000 population in 2007 (1). The incidence of brain abscesses is not known precisely but is estimated to be approximately 1 per 100,000 population (2,3). In contrast, the incidence of severe sepsis in the United States in 2007 was 303 cases per 100,000 population (4).
To establish bacterial CNS infections, pathogens must gain access either to the subarachnoid space (in the case of meningitis) or to the brain parenchyma (in the case of brain abscess). Many of the complex processes involved in the transition of pathogens from outside the host into the CNS have been elucidated, particularly for meningitis. Most cases of bacterial meningitis likely arise from bacteremia, which is caused by invasion of the bloodstream by the pathogen after colonization of the nasopharyngeal and intestinal mucosa. Similarly, in the pathogenesis of brain abscess, the hematogenous route is important in a substantial number of cases. However, the mechanisms by which pathogens gain access to the site of infection from the bloodstream likely differ substantially between meningitis and brain abscess. This is suggested by the fact that organisms typically causing bacterial meningitis (e.g., Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis) very rarely cause brain abscess, whereas pathogens typically found in brain abscess (e.g., aerobic and anaerobic Streptococcus species and Staphylococcus aureus) are rarely the cause of bacterial meningitis. Further elucidation of the molecular events leading to CNS invasion is likely to allow a better understanding for these differences.
Not all bacterial infections of the CNS are the result of bacteremia. This is most obvious when bacterial meningitis occurs as a consequence of infection of a cerebrospinal fluid (CSF) shunt or when a brain abscess results from breaching of the skull and meninges by trauma or a neurosurgical procedure. Similarly, in patients in whom brain abscess or meningitis originates from a focal infection in the vicinity of the brain (i.e., sinusitis, otitis media, dental abscess), contiguous spread rather than bacteremia represents the likely route by which the pathogen gains access to the CNS. Again, the pathogen causing the focal infection seems to determine whether the secondary infection is meningitis (in the case of S. pneumoniae or H. influenzae) or brain abscess (in the case of streptococci other than S. pneumoniae), suggesting pathogen-dependent differences in pathogenesis.
Multiple processes occur from the time a pathogen has reached the site of infection until the full manifestations of the disease have developed. These include the induction of cytokines and chemokines, activation of inflammatory mediators such as nitric oxide (NO), reactive oxygen species (ROS), or matrix metalloproteinases (MMPs), recruitment of white blood cells to the site of infection, and cytotoxic events. Many of these events have been analyzed in detail in bacterial meningitis, whereas less is known in the case of brain abscess.
This chapter focuses on the pathogenesis of bacterial meningitis and the subsequent development of brain damage. Aspects of the pathogenesis of brain abscess are discussed in detail in Chapter 31.
PATHOGENESIS OF BACTERIAL MENINGITIS
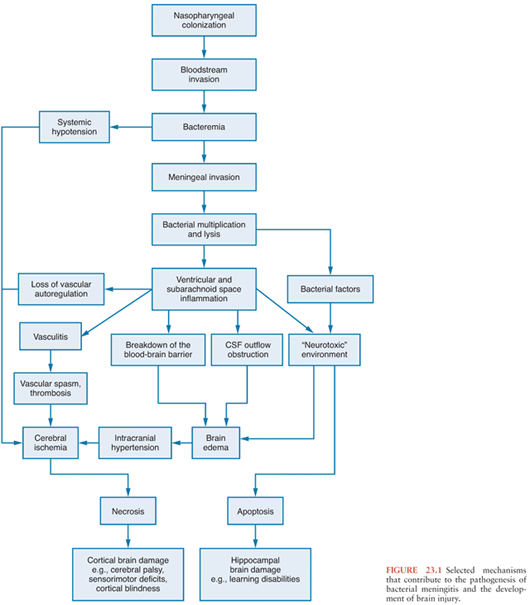
The development of bacterial meningitis progresses through four interconnected phases: (a) bacterial colonization and invasion of the host with subsequent infection of the CNS, (b) bacterial multiplication and induction of inflammation in the subarachnoid and ventricular space, (c) progression of inflammation with associated pathophysiologic alterations, and (d) damage to the CNS (Fig. 23.1).
Bacterial Invasion of the Host and Penetration of the Blood–Brain Barrier
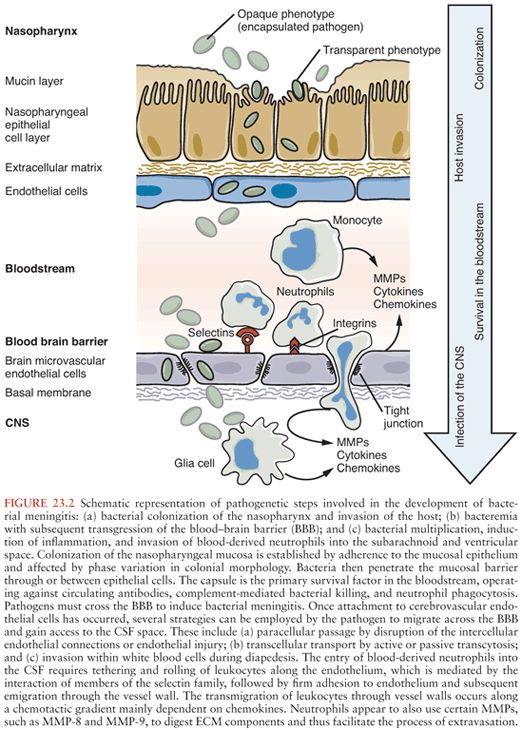
Figure 23.2 shows the pathogenic steps involved in the development of bacterial meningitis.
Colonization
Before a meningeal pathogen can cause invasive disease, it has to successfully colonize the host first. S. pneumoniae, N. meningitidis, and H. influenzae type B, which most frequently cause bacterial meningitis in humans, colonize the nasopharynx and are transmitted from person to person by the respiratory route. On the other hand, group B Streptococcus (GBS) (Streptococcus agalactiae), Escherichia coli, and Listeria monocytogenes, which cause meningitis at the vulnerable extremes of age and in immunocompromised persons, colonize the gastrointestinal tract and are transmitted through the oral, vaginal, or fecal-oral route.
Colonization of the nasopharyngeal mucosa is established by evasion of mucosal host defense mechanisms, for example, ciliary clearance and secretory immunoglobulin A (IgA), successful competition with other organisms in the aerobic environment of the upper respiratory tract, and adherence to the mucosal epithelium. Specialized surface components, such as components of the polysaccharide capsule, or pili, and the production of bacterial enzymes are crucial for the various steps necessary to establish colonization.
The mucociliary escalator is the main mechanical defense machinery of the respiratory tract. It constantly propels particles, including bacteria, to the outside. In order to gain access to the mucosa and establish themselves in the respiratory tract, S. pneumoniae, N. meningitidis, and H. influenzae type B have to overcome the mucociliary escalator. As exemplified by S. pneumoniae, there are several means by which bacteria may avoid being trapped and flushed away by mucus. The polysaccharide capsule of S. pneumoniae is almost exclusively negatively charged. This could increase electrostatic repulsion from the likewise negatively charged sialic acid residues of mucus (5). Additionally, deglycosylation of mucus by secreted exoglycosidases (e.g., neuraminidase A [NanA], beta-galactosidase B, etc.) may decrease its viscosity and lessen entrapment (6). Furthermore, pneumolysin, a major virulence factor of S. pneumoniae that is released by autolysis, inhibits ciliary beating (7,8). Pneumolysin has been shown to be necessary for successful colonization of the nasopharynx (9). Ciliostasis has also been documented in H. influenzae infection (10), whereas nasopharyngeal colonization of N. meningitidis entails cytotoxicity for ciliated epithelial cells (11).
Besides its mechanical action, respiratory mucus also harbors several soluble antibacterial agents. Secretory IgA antibodies inhibit microbial adherence and penetration into the mucosa. N. meningitidis, S. pneumoniae, and H. influenzae, as well as some members of the resident oral flora, secrete highly specific endopeptidases that cleave the heavy chain of human IgA1, including its secretory form (sIgA1), thus separating the monomeric antigen-binding fragments (Fab) from the secondary effector functions of the IgA1 antibody (Fc) (12). IgA1 proteases exist as three different classes of proteinases. IgA1 proteases of H. influenzae and N. meningitidis are genetically related serine proteinases, those of S. pneumoniae are metalloproteinases. Cleavage of IgA1 by proteases impairs specific mucosal immunity in the upper respiratory tract, thus allowing bacteria to colonize. IgA1 proteases may also contribute to the pathogenesis of invasive infections by leading to the coating of bacteria with Fab fragments, which mask epitopes and thus protect them from subsequent recognition by intact immunoglobulin (13). Lysozyme is an important component of the innate immune system and abundantly present in the saliva, airway fluid, and lysosomal granules of neutrophils. Its muramidase activity leads to hydrolyzation of the bacterial cell wall polymer peptidoglycan. Modification of peptidoglycan by N-deacetylation and O-acetylation and the production of lysozyme inhibitors are bacterial strategies to fend off lysozyme action (14). The gene coding for the enzyme responsible for N-deacetylation, peptidoglycan N-acetylglucosamine deacetylase A (PdgA), is present in S. pneumoniae and L. monocytogenes (15). Genes encoding for peptidoglycan O-acetyltransferases have also been found in S. pneumoniae (attenuator of drug resistance [adr]) and N. meningitidis (peptidoglycan acetylase A [pacA] and pacB) (16). For S. pneumoniae, it has been shown that expression of PdgA and Adr is necessary to provide lysozyme resistance and provide colonization advantage in the mouse model (17). Finally, apolactoferrin, the iron-depleted form of lactoferrin, is bacteriostatic or bactericidal for bacteria. The pneumococcal surface protein A (PspA) protects against the bactericidal effect of apolactoferrin, probably by binding to the active sites of apolactoferrin (18).
Bacterial adherence to epithelial cells is mediated by adhesins on the bacterial surface binding to epithelial cell receptors. Based on their structure, fimbrial and afimbrial adhesins can be distinguished in meningeal pathogens. Fimbriae (or pili) are filamentous structures, first identified in Gram-negative bacteria, that protrude from the outer cell membrane. Fimbriae are composed of several subunits that form rodlike structures and carry a specific adhesive subunit at their tip (19,20). Fimbriae have been associated with the early steps of adhesion mostly, but not exclusively, in Gram-negative meningeal pathogens. Several afimbrial adhesins have been identified in all meningeal pathogens and will be discussed with the respective pathogens.
Adherence of pneumococci to mammalian cells and pneumococcal virulence is facilitated by regulated expression levels of the polysaccharide capsule in a process called phase variation. Capsular phase variation of S. pneumoniae is marked by changes in colonial morphology from opaque to transparent and correlates with differences in virulence. The transparent, less capsulated phenotype is more capable of colonizing the nasopharynx, whereas the opaque phenotype shows increased virulence during systemic infections (21,22). Masking of underlying adhesion molecules by the polysaccharide capsule is a possible explanation for the inferior colonization capabilities of opaque strains. Adherence of S. pneumoniae in itself is thought to involve binding to glycoconjugates on the surface of the respiratory epithelium, although the exact mechanisms have not been completely elucidated (23). Recognition of these binding sites is enhanced by cleavage of the terminal sialic acid by NanA and other glycosidases (24,25). Further putative roles of NanA during the colonization process are its involvement in the formation of biofilm (26) and competition with other meningeal pathogens (27). A specific function as adhesin has been shown for the choline-binding protein A (CbpA; also called PspC or SpsA), which binds to the polymeric immunoglobulin receptor (pIgR) on human mucosal epithelial cells and complement factor H (23). Importance of CbpA has been demonstrated in an infant rat model where CbpA-deficient pneumococci were significantly restricted in their ability to colonize the nasopharynx (28). CbpA is a member of the family of choline-binding proteins. This family of proteins shares phosphorylcholine, a component of lipoteichoic and teichoic acids, as common cell wall anchor. Interestingly, phosphorylcholine also binds to activated platelet-activating factor receptor (PAFr) on epithelial cells and C-reactive protein (CRP) (29). Inflammation enhances this process, as expression of PAFr on lower respiratory tract epithelial cells, and thereby PAFr-dependent adhesion and transcytosis of S. pneumoniae, are increased by rhinovirus, acid, fossil fuel–derived particulate matter, and cigarette smoke (30). On the other hand, phosphorylcholine expression itself can be modulated by the pneumococcal phosphorylcholine esterase. This may allow the bacterium to limit CRP-binding and consecutive complement activation (23). Eventually, cell–cell adhesion molecules have been identified as important adhesion targets for viruses, bacteria, and parasites (31). For S. pneumoniae, it has been shown that pneumococcal surface antigen A (PsaA) attaches to E-cadherin, an adhesion receptor it shares with L. monocytogenes (32). CbpA and PsaA are produced by virtually all clinical isolates of S. pneumoniae and contribute to their virulence (33). It has also been shown in children that pneumococcal carriage and infection induce salivary and serum antibodies against NanA, CbpA, and PsaA, among other pneumococcal antigens (34,35). Also, levels of PsaA and NanA antibodies in the serum were inversely correlated to frequency of respiratory tract infections (35).
Primary adherence of N. meningitidis depends on the binding of type 4 pili on the bacterial cell surface to nonciliated epithelial cells. Thereby, N. meningitidis induces rearrangements of the cellular cytoskeleton leading to protrusion of microvilli and formation of cortical plaques in epithelial cells (10). Clusters of bacteria are then embraced by microvilli, increasing stability of the adhesion. Further physical proximity of the bacterial and epithelial cell surfaces accompanied by the loss of fimbriae and downregulation of capsule allows other adhesins to attach to their respective receptors (36). Most notably, opacity-associated (Opa) proteins, a family of phase-variable outer membrane proteins of N. meningitidis, bind to carcinoembryonic antigen-related cell adhesion molecules (CEACAM), cell surface–expressed heparan sulfate proteoglycans, and extracellular matrix (ECM) proteins (37). CEACAMs are further involved in the invasion process of N. meningitidis (discussed below). While fimbriae protrude from the polysaccharide capsule, the capsule interferes with the function of other adhesins of N. meningitidis (38,39). Downmodulation of the capsule or inducing the upregulation of adhesion receptors (e.g., CEACAMs) are proposed mechanisms by which invasive N. meningitidis overcome this disadvantage (38).
Although structurally different, fimbriae are also important adhesion molecules of H. influenzae (40). They mediate adhesion to mucin, ECM, and epithelial cells (41). Besides fimbriae, the protein Hsf is a major adhesin of H. influenzae. Hsf is anchored in the outer membrane of H. influenzae and forms fiber-like structures on the cell surface (41). Similar to N. meningitidis, the outer membrane protein P5 of H. influenzae binds to CEACAM-1 and intercellular adhesion molecule-1 (ICAM-1) (42,43).
E. coli and GBS are the main pathogens of neonatal meningitis but, like L. monocytogenes, may also cause meningitis in the elderly and immunocompromised persons. Transmission from the asymptomatically colonized mother or the environment can lead to the colonization of the intestinal tract of the infant, the first step in the pathogenesis of neonatal E. coli and GBS meningitis. The food-borne pathogen L. monocytogenes disproportionately affects pregnant women and is transmitted to the fetus through the placenta (44).
Similar to N. meningitidis, adherence of E. coli to host cells depends primarily on fimbriae. For example, the type I fimbriae in E. coli harbor the adhesive FimH subunits on their tip (45). FimH binds mannose-containing residues on the host cell and has been implicated in the tissue tropism of pathogenic E. coli (46). Additionally, type S fimbriae are expressed by E. coli that have been isolated in neonatal meningitis and sepsis cases (47). Another heterogenous family of adhesins that may be assembled into fimbriae-like structures are the Afa/Dr adhesins that have been reported to adhere to the decay-accelerating factor (DAF) expressed on cells exposed to plasma, complement proteins, CEACAMs, and type IV collagen (19). The expression of adhesins on E. coli differs between the different pathogenic and nonpathogenic subtypes and likely determines the organ tropism. Accordingly, the majority of E. coli isolates found in neonatal meningitis exhibits the capsular serotype K1 and a limited repertoire of different O antigens (48). The reason for the neurotropism of these specific serotype combinations is not yet completely understood, although the K1 capsule seems to be the decisive factor for meningitis development (49).
Akin to neonatal E. coli meningitis, epidemiologic analysis of GBS in neonates with GBS meningitis shows a predominance of the capsular serotype III and the clonal complex ST-17 (50). Recently, a novel virulence factor has been described in bacteria belonging to the ST-17 complex. The hypervirulent GBS adhesin is anchored to the cell wall and in vitro significantly enhances adhesion to intestinal epithelial cells, resulting in increased colonization of the intestine in the in vivo mouse model (50). Other adhesins involved in the attachment of GBS to the gastrointestinal epithelium primarily bind to components of the ECM. For example, the cell surface protein SepB binds to fibronectin but also mediates cleaving of complement factor C5a. Attachment to laminin is mediated by the adhesin Lmb, whereas the surface-anchored protein FbsA binds to fibrinogen (51). Finally, fimbriae have also been involved in GBS adherence and biofilm formation (52).
For adherence and internalization of L. monocytogenes at the intestinal mucosa level, the interaction of the internalin A (InlA) with the cell-junction protein E-cadherin is paramount (53,54). E-cadherin is normally located below the tight junction and not available as a receptor to intraluminal bacteria. However, it has been demonstrated that E-cadherin is present on the luminal surface of mucus-secreting goblet cells and may be temporarily accessible at the tips of intestinal villi due to constant shedding of dead enterocytes (55,56). Whereas adherence to the intestinal epithelium is important for the pathogenesis of L. monocytogenes in the elderly and immunocompromised persons, transmission of L. monocytogenes to the neonate occurs via the transplacental route. Histopathologic analysis of placentas from women with fetoplacental listeriosis and in vitro analysis indicate that L. monocytogenes targets E-cadherin on the surface of syncytiotrophoblasts in a similar way as in the intestine (57).
Invasion of the Bloodstream
The functional integrity of the respiratory tract mucosa is critical in protecting the host from bacterial invasion. Viral infections of the respiratory tract may lead to a decrease in ciliary function and associated mechanical clearance of bacteria, upregulation of different host cell receptors involved in bacterial adherence, and alteration of host immune response (58,59), and are associated with an increased risk of invasive bacterial disease. Epidemic outbreaks of meningitis due to N. meningitidis in the sub-Saharan “meningitis belt” coincide with the dry periods of the year, when the protective barrier of the mucous membrane is compromised (60).
Bacteria can penetrate the mucosal barrier through or between epithelial cells, and both routes seem to be employed depending on the pathogen. Presence of a capsule or sialylated lipooligosaccharides, factors that are instrumental for the survival of meningeal pathogens in the bloodstream, hinders the process of transcytosis (10,61). Most of the identified mechanisms by which these pathogens directly invade human nasopharyngeal mucosal cells take advantage of a ubiquitous cellular endocytosis mechanisms. In clathrin-mediated endocytosis, a ligand-receptor interaction initiates the formation of a clathrin-coated pit, which eventually forms a vacuole that engulfs particles (62,63). Additionally, actin rearrangements are induced and further enhance bacterial internalization. On the other hand, paracellular migration of bacteria might be facilitated by toll-like receptor (TLR)–dependent downregulation of tight junction components (64) or degradation of intercellular junctions mediated by bacterial enzymes or bacteria-bound plasmin (65–68). So far, several host cell factors have been identified as targets of bacterial invasion mechanisms.
The transmembrane receptor pIgR is necessary for transport of IgA and IgM across epithelial cells. After binding of the immunoglobulins at the basolateral surface of the epithelial membrane, the pIgR-immunoglobulin complex is taken up into endosomes and transported across the epithelium in vesicles to the apical mucosal surface, where it is released into mucosal secretions (69). S. pneumoniae is capable of exploiting this mechanism for its own transcytosis across mucosal cells. As previously mentioned, binding of S. pneumoniae to human pIgR is mediated by CbpA. This interaction allows S. pneumoniae to hijack the recycling pathway of pIgR from the apical to the basal cell surface by retrograde transport, thereby facilitating transmucosal invasion of pneumococci (70,71).
The CEACAM proteins, a family of proteins involved in intercellular adhesion, have been identified as target of adhesion molecules of N. meningitidis, H. influenzae, and E. coli (72). Of the CEACAMs that have been associated with bacterial binding, CEACAM-1 is most widely distributed on different human cell types. Other CEACAMs that have been shown to bind to Opa of N. meningitidis are CEACAM-3, CEACAM-5, and CEACAM-6 (73). Interestingly, binding to CEACAM-1 does not only allow N. meningitidis to adhere and invade epithelial cells by endocytosis (74) but may also downregulate the host immune response on several levels (72). On the other hand, binding of N. meningitidis to CEACAM-3 on neutrophils has been shown to enhance engulfment and killing of bacteria (73).
E-cadherin is the cell surface protein responsible for establishment of adherens junctions between neighboring cells (31). In the intestine, the interaction of listerial protein InlA with E-cadherin leads to the rearrangement of the actin skeleton of the host cell and ensuing engulfment of the bacteria into a vacuole that is transported to the basolateral cell surface (56). Persistence in the vacuole until release into the lamina propria is in contrast to intracytosolic release and replication of L. monocytogenes in other human cells. Although interaction of InlA with E-cadherin is also involved in the invasion process of L. monocytogenes in the placenta, additionally, binding of InlB to its host receptor Met is necessary (75).
The exact mechanisms by which E. coli K1 and GBS invade human mucosal cells have not been elucidated. One adhesin/invasin that has been identified in E. coli K1 is the Hek outer membrane protein, which mediates binding to proteoglycan on the epithelial cell surface (76). Additionally, a role for outer membrane protein A (OmpA) and IbeB, better studied in BBB crossing, has been suggested (76). Altogether, akin to other meningeal pathogens, this points to an endocytosis-like process which is enabled by rearrangement of the epithelial cell cytoskeleton (77).
Between epithelial cells and mesenchymal cells lies the basement membrane, providing a final mechanical barrier before the invading pathogen can disseminate throughout the host. Attachment to ECM components is an important property of bacterial infection pathogenesis, and ECM adhesins have been identified in most meningeal pathogens (e.g., PavA of S. pneumoniae [23], Opc of N. meningitidis [78], fimbriae of H. influenzae [79], and ScpB of GBS [51]). To cross the basement membrane, meningeal pathogens have devised several strategies: degradation of the basement matrix by secretion of proteolytic enzymes, use of the host’s plasminogen-plasmin system, and possibly crossing of the basement membrane in phagocytic cells. Hyaluronidase is an endoglycosidase that degrades hyaluronic acid, a component of the ECM. S. pneumoniae strains demonstrate a strong correlation between hyaluronidase activity and the capacity to induce meningitis (80). Intranasal instillation of pneumococci together with hyaluronidase was followed by meningitis in 50% of inoculated mice, whereas mice inoculated without hyaluronidase failed to develop meningitis (65). Plasminogen is the key proenzyme of the fibrinolytic system, which after activation becomes the broad-spectrum protease plasmin. Although fibrin is its main substrate, plasmin can cleave several components of the basement membrane and ECM (81). Binding of plasminogen has been shown for N. meningitidis, S. pneumoniae, and H. influenzae (82) and enhances invasiveness of S. pneumoniae and H. influenzae (67,83,84). Phagocytic cells targeting colonizing pathogens on the mucosal surface move freely between the mucosal surface and the intraluminal space of the mucosal microvasculature. This movement of phagocytic cells can be exploited by bacterial pathogens that are able to survive in phagocytic cells (85). For L. monocytogenes, it has been shown that dendritic cells are an important transport vehicle from the mucosa to lymphatic organs (86).
Intravascular Survival
Once in the bloodstream, meningeal pathogens are immediately attacked by the host immune defense. To establish successful bacteremia, meningeal pathogens need to overcome the different effector systems of the immune defense. Clearance of the classical meningeal pathogens S. pneumoniae, H. influenzae, and N. meningitidis relies on antibody-mediated opsonization followed by activation of the complement system. In the case of S. pneumoniae and H. influenzae, this is followed by phagocytosis and finally intracellular killing, whereas the membrane attack complex is important for control of N. meningitidis (87). In neonates with E. coli and GBS meningitis, immune defense relies especially on innate immunity, with the complement system being the first line of defense. The importance of the complement system for the clearance of meningeal pathogens is underlined by the fact that patients with impaired complement activation (i.e., sickle cell disease, anatomical or functional asplenia, or polymorphisms in the complement components) are predisposed to invasive pneumococcal infections, whereas patients with deficiencies in the terminal complement components are prone to invasive infections with N. meningitidis (88–91).
The complement system may be activated by the classical pathway, the mannose-binding lectin (MBL) pathway, or the alternative pathway, all converging at the deposition of activated complement factor C3b on the pathogen surface, which catalyzes the downstream effects—opsonization and phagocytosis or bacteriolysis—of the complement system (87). The meningeal pathogens have developed several mechanisms to evade deposition of C3b on the cell surface. The polysaccharide capsule, although hindering adherence to the mucosal surface and entry into epithelial cells, is the primary survival factor in the bloodstream, operating against circulating antibodies, complement-mediated bacterial killing, and neutrophil phagocytosis (49). Accordingly, encapsulation is a shared feature of the major hematogenous meningeal pathogens (H. influenzae, N. meningitidis, S. pneumoniae, E. coli K1, and GBS). Sialic acid, which is present on most human cells and blocks activation of complement, is also found in the capsule of meningeal pathogens, for example, GBS (type III), E. coli (K1), and meningococci (groups B and C). Thereby, the polysaccharide capsule may prevent complement-mediated bacteriolysis (92). Also, in S. pneumoniae, the capsule is thought to interfere with the activation of the classical complement pathway by limiting binding of immunoglobulin and CRP to subcapsular antigens (93). Binding of immunoglobulin may further be hindered by the action of IgA1 proteases. IgA1-proteases cleave IgA1 in the hinge region and liberate monomeric Fab-a fragments, which block the access of intact immunoglobulin G (IgG) or immunoglobulin M (IgM) to the pathogen (94). Several host defense mechanisms counteract the antiphagocytic activity of the bacterial capsule. Most effective are antibodies against capsular components, which permit optimal opsonization of bacteria and ensure efficient phagocytosis by polymorphonuclear (PMN) leukocytes and macrophages. The efficacy of this mechanism is underscored by the more than 95% decrease of H. influenzae type b meningitis after the introduction of conjugate vaccines, which induce high anticapsular antibody titers even in young children (95).
The first step in the classical complement pathway is the association of C1-complex to antibodies bound to the bacterial surface. The pneumococcal protein PspA has been shown to limit this step (96). C1-complex (classical complement pathway) or MBL-associated serine protease 2 (MBL pathway) then activate the C3-convertase C4b2a, which in turn activates C3 to C3b. As with any enzyme cascade, activation of the complement system needs to be balanced carefully to avoid damage to host cells. Regulators of complement activation include C4-binding protein (C4BP), factor H, and DAF, which accelerate the dissolution of C3-convertases (97). E. coli K1, N. meningitidis, S. pneumoniae, GBS, and H. influenzae may all inhibit the classical and MBL complement pathway by binding of C4BP (98,99). Also, S. pneumoniae, N. meningitidis, and GBS can bind factor H and downregulate the alternative complement pathway (100–102). In particular, meningococcal factor H–binding protein has received considerable attention because this molecule is a key component of group B meningococcal vaccines (103,104). The result of C3 activation is the generation of C5 convertase. C5 convertase cleaves C5 into C5a, a strong chemoattractant and anaphylatoxin, and C5b, member of the membrane attack complex (MAC). GBS express an endopeptidase that cleaves C5a (105). The final step of complement cascade is the generation of the MAC, which mainly affects Gram-negative pathogens. Vitronectin regulates this last step of the complement cascade by interfering with MAC assembly. H. influenzae and N. meningitidis have been shown to bind vitronectin through Hsf and Opc, respectively, and thereby limit bacteriolysis (106). Although evasion of the complement system also impedes opsonic phagocytosis, the polysaccharide capsule of the classical meningeal pathogens additionally allows for direct evasion of bacterial uptake and killing by professional phagocytes (107,108).
In contrast, the intracellular pathogen L. monocytogenes depends mainly on its ability to evade intracellular killing in the phagolysosome to evade the host immune system. After uptake into the cell, L. monocytogenes can rupture the vacuole through the action of two phospholipases and listeriolysin and escape into the cytosol (109). Concurrently, N. meningitidis and S. pneumoniae have been shown to escape the oxidative burst in leukocytes by guarding themselves against ROS by the production of glutathione (110,111). Another important virulence factor of L. monocytogenes is the actin assembly–inducing protein ActA, which catalyses polar actin assembly and autophagy escape, thereby enabling intracellular motility and spread to adjacent cells (109). Although bacterial clearance by phagocytosis is an important early step in listeriosis, the ability of L. monocytogenes to survive intracellularly in macrophages allows it to travel to target organs, thereby evading innate immune mechanisms.
In summary, the intricate defense mechanisms of bacteria against the host immune system only need to safeguard the survival of one (or a few) organism(s), which may then gain access to the subarachnoidal space and cause meningitis (112).
Meningeal Invasion
To enable efficient neuronal signaling, the CNS is a tightly regulated space protected from its surroundings by different barriers. The boundary to the blood is provided by the BBB, the blood–cerebrospinal fluid barrier (BCSFB), and the arachnoid (113). The BBB is composed of brain endothelial cells, closely connected through adherens and tight junctions; a basal membrane; astrocytic and microglial processes; and pericytes. The close interconnection of endothelial cells, low pinocytic activity, and specific transport mechanisms allow for a selective permeability (113). Bloodborne pathogens must cross the BBB or the BCSFB from the bloodstream to induce bacterial meningitis. Clinical observations and experimental studies demonstrate a relationship between the magnitude of bacteremia and the development of meningitis for most meningeal pathogens (89,114,115). However, high bacteremia alone is not sufficient for the development of bacterial meningitis, but expression of selective bacterial adhesion and invasion factors is necessary (116).
Attachment to the brain microvascular endothelial cells is facilitated by receptors for meningeal pathogens found on the endothelium in cerebral capillaries and the choroid plexus. The laminin receptor, a cell surface protein involved in binding to ECM components, has been suggested as common adhesion target for S. pneumoniae, N. meningitidis, and H. influenzae and also supports invasion of cytotoxic necrotizing factor 1 (CNF1) expressing E. coli K1 (117,118). Adhesion to brain endothelial cells via the laminin receptor is mediated by CbpA in pneumococci, outer membrane proteins PilQ and PorA in meningococci, and the porin OmpP2 in H. influenzae (117). Further, fimbriae are important structures in bacterial adhesion to human cells (see earlier discussion). Fimbriae are essential for binding of N. meningitidis to brain endothelial cells (119). Binding of E. coli K1 to brain endothelial cells is also mediated by FimH on type 1 fimbriae binding to CD48 and OmpA association with glycoproteins on the cell surface (116). For GBS as well, several adhesins are required for binding to brain endothelial cells, including the pilus tip adhesin PilA (120–122).
Once attachment has occurred, pathogens may employ several strategies to migrate across the BBB and gain access to the CSF space. These include (a) transcellular traversal by transcytosis, (b) paracellular passage by disruption of the intercellular endothelial connections or endothelial injury, and (c) invasion within white blood cells during diapedesis. Causative organisms of bacterial meningitis have mainly been shown to migrate into the CSF by transcellular or paracellular migration (116). However, L. monocytogenes also accesses the CSF space inside leukocytes (123).
Direct invasion of endothelial cells by meningeal pathogens involves ligand-receptor interactions between bacteria and host cells and rearrangement of the endothelial actin cytoskeleton, which ultimately leads to the uptake of the pathogen in a vacuole and transcellular transport (116,124). For S. pneumoniae, the interaction of phosphorylcholine with PAFr and expression of NanA are important for invasion of the BBB (125). PAFr is a transmembrane receptor expressed by several different cell types across the human body and involved in the pathogenesis of different inflammatory diseases, atherogenesis, embryo implantation, and CNS signaling (126). PAFr is a G protein–coupled receptor and the ligand-receptor complex can be taken up into a vacuole that either recycles back to the cell surface or fuses with lysosomes to terminate signaling (126). Similarly, pneumococci bound to PAFr via phosphorylcholine are taken up into vacuoles but may then avoid fusion of the vacuole with lysosomes. Instead, they are transported to the basolateral surface of the endothelial cell and released out of the cell (127). Likewise, IbeA and CNF1 proteins are involved in the invasion of brain endothelial cells by E. coli K1, ultimately leading to the uptake of E. coli into a vacuole (116,128). An important aspect of transcytosis of meningeal pathogens is the ability of bacteria to avoid fusion of the vacuole with lysosomes as demonstrated for E. coli K1 (129).
Interaction of bacterial adhesins with ECM proteins may enhance invasion of meningeal pathogens. In vitro evidence has been brought forward for the exploitation of the integrin receptor–mediated mechanism of ECM protein internalization by N. meningitidis and GBS (122,130). On the other hand, for N. meningitidis, experimental evidence indicates that the formation of cortical plaques, induced by microcolonies on brain endothelial cells, and the ensuing reorganization of the intracellular cytoskeleton lead to the opening of intercellular junctions followed by paracellular penetration of the BBB (119). Receptor-mediated signaling as well as local inflammation with recruitment of leukocytes may further modify tight junctions and allow bacteria to penetrate paracellularly (119,122).
Transcellular invasion of brain endothelial cells by L. monocytogenes is thought to be mediated by InlA and InlB in similar fashion as in the intestine and placenta, although conflicting experimental evidence exists (123,131). Additionally, the effective intracellular trafficking of L. monocytogenes across the BBB in phagocytic cells has been shown in a mouse model (132,133).
In humans, invasion of the CSF by bacteria can occur independently of bacteremia. This happens typically when focal infections occur in structures in immediate proximity to the brain (e.g., otitis media, mastoiditis, sinusitis) or when the integrity of the skull and meninges surrounding the brain is disrupted (e.g., malformations, trauma, neurosurgery). In a case series of 87 patients with pneumococcal meningitis, more than half of the patients had radiologic signs of otitis or sinusitis (134), whereas in a larger observational study, otitis and sinusitis were reported as predisposing conditions in 25% of patients (135). In a model of pneumococcal respiratory tract infection, meningitis occurred without detectable bacteremia, suggesting that pneumococci are able to invade the brain by bypassing the bloodstream (136). Similarly, a galU mutant and its parent pneumococcal strain both caused meningitis following otitis media infection in gerbils, despite the mutant’s impaired ability to disseminate to the bloodstream following infection (136).
For L. monocytogenes, S. pneumoniae, and N. meningitidis, retrograde access to the CNS via the neural route has also been documented (123,137,138). However, for L. monocytogenes, this mechanism is likely only important in ruminants. These observations support the notion that meningeal pathogens can gain access to the subarachnoid space by several routes. The relative importance of hematogenous versus nonhematogenous routes of infection in humans is not exactly known.
Meningeal Infections Associated with Foreign Bodies
Pathogenesis of foreign body–associated meningeal infection differs in several important aspects from bacterial meningitis described earlier. First, at the time of foreign body insertion and due to local tissue damage, access of bacterial pathogens to the CSF space is facilitated. Second, bacteria may organize on the foreign body in form of biofilm, thereby defying the host’s immune system and systemically administered antimicrobials. Finally, foreign body-associated meningitis frequently brings forth only low-grade inflammation, with fever, irritability, and shunt malfunction being the most prevalent clinical findings (139,140). CSF shunt infections develop into clinical ventriculitis or meningitis only in approximately 30% of patients. In patients with hydrocephalus, CSF shunts convey CSF from the lateral cerebral ventricles to the peritoneum or right cardiac atrium or transiently to an external collection system. Shunt infection is one of the main complications associated with CSF shunts. The reported incidence of CSF shunt infection is in the range of 5% to 15% (141–144), with pediatric patients being more affected than adults (143). Shunt infections occur primarily as an early postoperative event, which manifests itself within a few days or weeks after surgery (139,142,143). Due to the close association with the operative procedure and the prevalence of bacteria belonging to the skin flora, intraoperative inoculation is thought to be the most likely cause of infection in early CSF shunt infection. However, late shunt infections can occur several years after implantation, likely due to hematogenous infection or associated with intraabdominal infection (139,143). Shunt colonization with bloodborne pathogens such as H. influenzae or S. pneumoniae has also been found, and one study reported five cases of meningitis due to N. meningitidis, H. influenzae, and S. pneumoniae out of a series of 289 patients with CSF shunt followed for 10 years (145). Nevertheless, most shunt infections are caused by staphylococci, in particular S. epidermidis, which causes half of all cases (143,144,146). The characteristic feature of CSF shunt infection is the adherence of bacteria to the inner surface of the shunt tubing. Insertion of a CSF shunt leads to local inflammation and deposition of ECM proteins, that is, fibronectin or fibrinogen, on the shunt surface (147). Binding of ECM proteins via fibrinogen-binding protein (148), fibronectin-binding protein (149), and vitronectin-binding proteins (150) enhances adherence of S. epidermidis to the shunt surface. In an experimental model of catheter-associated infection, presence of a foreign body reduced the number of bacteria needed to establish infection (151). Once attached to the shunt surface, S. epidermidis rapidly organizes into a biofilm, characterized by bacteria embedded in a self-generated polysaccharide matrix (152,153). Although shunt infection does elicit an inflammatory response in the CNS (154), staphylococci forming a biofilm are relatively protected from the host immune defense mechanisms (151,155,156). Additionally, bacteria organized in a biofilm have a greatly increased resistance to systemic antimicrobial therapy. This is likely due to the diversity of metabolic states of bacteria inside the biofilm combined with the physical protection by the extracellular polysaccharide matrix (153,157). Penetration of most antimicrobials through the BBB does not suffice to reach the necessary concentrations needed to kill bacteria inside a biofilm (158), therefore intraventricular administration of antibiotics and shunt removal are frequently necessary to treat CSF shunt infection.
The pathogenesis of Gram-negative CSF shunt infection is not well understood. Retrograde infection in which bowel perforation may lead to distal colonization of the shunt with subsequent retrograde migration to the ventricles is frequently suggested as the most likely mechanism for Gram-negative shunt infections.
The several times higher risk of patients after cochlear implantation to develop bacterial meningitis was first appreciated in 2002. Depending on the type of cochlear implant used, there was a 16 to 30 times higher incidence in bacterial meningitis than in the general U.S. population (159). S. pneumoniae accounted for 60% of the cases with known etiology (159). Incidence of bacterial meningitis in this special population has since then decreased, but it is still at least twice as high as in the general population (160). Cochlear implants are designed for people with profound bilateral sensorineural deafness in whom the auditory nerves are intact but hearing aids are ineffective. In animal studies, the insertion of an electrode array into the scala tympani reduced the number of bacteria needed to induce meningitis in animals challenged with S. pneumoniae by the hematogenous or otogenic route, whereas sole cochleostomy surgery did not (161). The exact impact of the foreign body in this setting has not been elucidated. To reduce the risk of post–cochlear implantation meningitis, it is suggested that patients are thoroughly evaluated for accompanying inner ear malformations, which may increase the risk for CSF leaks, and completion of their vaccination status. At surgery, measures should be taken to minimize communication between the middle and inner ear, as this is thought to be the mode of bacterial entrance in a sizable portion of meningitis cases in patients with cochlear implant (162).
Bacterial Multiplication in the Cerebrospinal Fluid
Host defense mechanisms in the CNS are tightly regulated. Peripheral immune cells and plasma proteins are largely excluded from the brain parenchyma by the BBB and BCSFB, and the brain parenchyma additionally features anti-inflammatory properties. Therefore, under physiologic conditions, microglia and astrocytes provide basic immune functions in the brain (recognition of pathogen-associated molecular patterns [PAMPs], initiation of inflammation), whereas peripheral leukocytes cross the BBB only in low numbers and stick to the perivascular space of CNS microvessels (163). In bacterial meningitis, the perivascular space and the meninges are the interface where cellular recognition of PAMPs and danger-associated molecular patterns elicits a strong inflammatory reaction and leads to the recruitment of peripheral immune cells to the CNS (164,165). In contrast to the peripheral blood, where cellular and soluble factors of the innate immune system are present in abundance and may act immediately on bacterial challenge, innate immune defense in the perivascular and meningeal space needs to be scaled up first (166,167). This delay in immune activation favors the survival of pathogens once they have reached the CSF. Bacteria can multiply within the CSF almost as efficiently as in vitro (168), reaching titers of up to 109 CFU/mL, spread over the entire surface of the brain and spinal cord, and extend into the Virchow-Robins space along penetrating vessels. Interestingly, presence or absence of leukocytes in the CSF does not affect bacterial multiplication in experimental meningitis models (169,170). In contrast, the complement system has been found to be instrumental to the control of bacterial multiplication in the CSF, supporting leukocyte influx, cytokine expression, and bacterial phagocytosis (171,172). Consistent with this, negative outcome in bacterial meningitis has been associated with depletion of complement factors in the CSF (166,173).
Gene expression analyses of S. pneumoniae have shown that, with exception of capsule genes, classical virulence genes (i.e., encoding for pneumolysin, autolysin, and pyruvate oxidase) are downregulated in the CSF (174–176). In contrast, upregulation of genes involved in amino acid acquisition/synthesis and energy metabolism has been documented in S. pneumoniae growing in CSF due to the scarcity of nutrients in the CSF (175,176).
Induction of Inflammation
Autolysis or exposure of bacteria to lytic antibiotics in the CSF leads to the release of subcapsular components (fragments of cell wall, lipopolysaccharide, teichoic and lipoteichoic acid, peptidoglycans, bacterial DNA, and other cytosolic factors) that trigger the inflammatory response in the subarachnoid space. Different cells from the tissues lining the CSF (ependymal and endothelial cells, perivascular macrophages) and brain-resident cells (mostly microglia, but also astrocytes and neuronal cells) are able to recognize these bacterial components. Distinct and often overlapping sets of specialized extracellular (i.e., TLRs) or intracellular (i.e., NOD-like receptors [NLR]) receptors are used by these cells to sense the major meningeal pathogens (177–179). Genetic variations related to these receptors are associated with negative outcome during bacterial meningitis (180–182). In response to bacterial stimuli, cells equipped with these specialized receptors produce cytokines, chemokines, and other proinflammatory molecules, leading to the recruitment of leukocytes to the site of infection (183,184).
Cytokines and Chemokines
A large variety of cytokines and chemokines have been detected in the brain parenchyma or the CSF during bacterial meningitis in patients or in experimental models (185–188). The pattern of cyto-/chemokines produced in response to the different meningeal pathogens has been shown to vary in in vitro and in experimental infection models. These differences are presumably related to pathogen-specific activation of pattern recognition receptors (189–192). They may contribute to the differences in outcome observed in patients affected by the different pathogens.
Early in the course of bacterial meningitis, tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6 are released, which then trigger, often in synergy, a cascade of inflammatory mediators, including other cytokines, chemokines, platelet-activating factor (PAF), antimicrobial peptides, prostaglandins, MMPs, NO, and ROS. The expression of TNF-α and IL-1β is detected first in the ependyma and the meninges and later in the parenchyma in experimental meningitis (194).
Increased CSF concentrations of TNF-α, IL-1β, IL-6, IL-8, and IL-10 are characteristic for bacterial meningitis. These cytokines are predominantly proinflammatory with the exception of IL-10, which downmodulates the production of TNF-α and other proinflammatory cytokines (194–196).
High concentrations of TNF-α in CSF were detected within 3 hours after injection of live H. influenzae in an experimental meningitis model (197). Intracisternal injection of S. pneumoniae led to a peak concentration of TNF-α in CSF approximately 12 to 24 hours after infection, with persistently elevated levels for at least 24 hours (198,199). The sustained TNF-α activity in the CSF may be explained by continuous stimulation by products released from bacteria in the CSF or by a positive feedback loop in the inflammatory cascade (198). TNF-α leads to nuclear factor-κB (NF-κB) activation, which regulates the expression of many proinflammatory genes (e.g., IL-1, TNF-α, IL-6, IL-8, macrophage inflammatory protein-1, inducible NO synthase [iNOS], cyclooxygenase-2, ICAM-1) (200,201).
Administration of TNF-α into the CSF results in pathophysiologic changes characteristic of bacterial meningitis, including BBB breakdown, neutrophil influx, and increase of cerebral metabolism, oxygen consumption, and cerebral blood flow (CBF), which in part are mediated through cyclooxygenase activity (202–207). Antibiotics cause rapid lysis of microorganisms and an associated brisk release of bacterial cell wall products, resulting in significantly higher TNF-α concentrations in CSF shortly after initiation of antimicrobial therapy (208,209).
The proinflammatory cytokine IL-1β is released by mononuclear phagocytes, glial cells, and endothelial cells in the CNS. Upon recognition of intracellular pathogens, the precursors of IL-1β or IL-18 are activated by caspase-1 in the inflammasome complex (179, 210–212). IL-1β is present in CSF samples of patients with bacterial meningitis, and its concentration is significantly correlated with inflammatory parameters, TNF-α concentrations, and adverse disease outcomes (213). IL-1 appears in the CSF of rats as early as 30 minutes after intracisternal injection of H. influenzae lipooligosaccharide (214) and 6 to 12 hours after injection of live S. pneumoniae in infant rats (199). When injected directly into the CSF of rabbits, IL-1 triggers a meningeal inflammation without detectable TNF-α activity, suggesting an independent mechanism of action, whereas simultaneous administration of TNF-α and IL-1 results in a synergistic increase in leukocyte influx into the CSF. TNF-α and IL-1 induce other secondary cytokines such as IL-6, IL-8, and IL-10 (213).
IL-6 is produced by monocytes, endothelial cells, and astrocytes, essentially in response to IL-1β. IL-6 is present in the CSF during meningitis later than TNF-α and IL-1β and remains present longer than the latter cytokines. Although IL-6 can be detected in the CSF of patients with bacterial meningitis, its presence is not correlated with any of the indices of meningeal inflammation or with severity of disease (215). IL-6 has a predominantly proinflammatory effect and is a potent inducer of acute-phase proteins, fever, leukocytosis, and activation of the complement and clotting cascades (216). Some antiinflammatory effects of IL-6 include inhibition of TNF-α and IL-1β production in vitro and induction of IL-1 receptor antagonist (217). Accordingly, knockout mice lacking IL-6 display an increase in pleocytosis and in the inflammatory response (218).
IL-8 (CXCL8) was the first identified member of a large family of chemokines shown to regulate the migration of leukocyte subsets toward the CNS during bacterial meningitis (187). Similar to most chemokines, its primary action is to activate and attract leukocytes, mainly neutrophils, to sites of inflammation. Cells shown to produce IL-8 upon stimulation with TNF, IL-1, or bacterial products include monocyte-macrophages, neutrophils, endothelial cells, astrocytes, microglia, and neurons. IL-8 is present in the CSF of patients with bacterial meningitis (187) and enhances neutrophil adhesion to endothelial cells, a prerequisite for the invasion of leukocytes into the brain. Belonging to the same chemokine subfamily as IL-8, the concentrations of CXC5 (ENA-78) and CXCL-1 (GRO-α) increase in the CSF of patients with bacterial meningitis, inducing neutrophil chemotaxis (119). To a lesser extent, another subfamily of chemokines, including monocyte chemoattractant protein-1 (MCP-1, CCL2), MIP-1α (CCL3), and MIP-1β (CCL3) is upregulated in the CSF of patients with bacterial meningitis and attracts mononuclear leukocytes (220).
IL-10 is an antiinflammatory cytokine that inhibits the production of TNF-α, IL-1, IL-6, and IL-8 in vitro and attenuates brain edema during meningitis. High levels of IL-10 have been found in the CSF of patients with bacterial meningitis, and in rabbits with experimental meningitis, administration of IL-10 downmodulated subarachnoid space inflammation (221). In an experimental rat model of pneumococcal meningitis, systemically, but not intrathecally, administered IL-10 reduced CSF pleocytosis and the concentration of proinflammatory cytokines (222). The inflammatory responses of IL-10 gene–deficient and wild type mice were compared in a mouse model of meningitis induced by intranasal inoculation of S. pneumoniae. Although antibacterial defense and survival were not influenced, the absence of IL-10 was associated with an increased immune response, as reflected by a more pronounced inflammatory subarachnoid infiltrate and higher concentrations of proinflammatory cytokines and chemokines in brain tissue (223). In experimental neonatal E. coli meningitis in mice, IL-10 favors the immune response by increasing the phagocytic activity of immune cells to clear the pathogen more rapidly and reduce the inflammatory response (224). In addition to IL-10, transforming growth factor-β (TGF-β) has also been shown to have antiinflammatory properties in experimental meningitis by reducing cerebral edema, intracranial pressure (ICP), and CBF (225). Conversely, by specifically blocking TGF-β signaling on neutrophils and macrophages, neutrophil influx and bacterial clearance were increased in experimental pneumococcal meningitis, which resulted in less secondary brain damage and improved survival (226).
Matrix Metalloproteinases and Related Proteases
The MMPs comprise endopeptidases that serve as effectors of cell migration, tissue remodeling, and cytotoxicity by degradation of ECM components. MMPs are synthesized as inactive zymogens and can be activated by conformational changes that disrupt a Zn2+ binding cysteine switch. MMPs not only function as effectors of tissue remodeling but also interact with the cytokine network. Cytokines such as TNF- α, IL-1, and IL-2 modulate the expression and regulation of MMPs. In return, MMPs and related metalloproteinases can act as sheddases or convertases as they transform membrane-bound cytokines, cytokine receptors, and adhesion molecules to their soluble forms. TNF-α converting enzyme (TACE/ADAM-17) is a member of the ADAM (a disintegrin and metalloproteinase) family of membrane proteins and is a highly efficient sheddase of TNF-α and TNF receptors. These have an integral role in the network of MMPs and cytokines (227).
Most MMPs are expressed only on demand through the action of cytokines, eicosanoids, growth factors, and components of infectious pathogens, among others. The most prominent of these inducers are the proinflammatory cytokines IL-1 and TNF-α. MMP-2, in contrast to other MMPs, lacks a classic promoter sequence and is constitutively expressed. Tissue inhibitors of metalloproteinases (TIMPs), the specific endogenous inhibitors of MMPs, form complexes with pro- and active forms of MMPs and inhibit the enzymatic activity. Similar to MMPs, TIMPs are also regulated by a network of different signaling molecules (228).
MMPs appear to play a central role in the development of bacterial meningitis (e.g., breakdown of the BBB, intrathecal production of cytokines, and accumulation of blood-derived leukocytes in the CSF) (229,230). Levels of MMP-1, -3, -7, -8, -9, and -10 have been found to be upregulated in the CSF of patients with bacterial meningitis, whereas TIMP-1 and TIMP-2 may be slightly upregulated. Interestingly, in the same patients, TIMP-4 was downregulated in the CSF (231–233). Immunohistochemical analysis of the brain tissue of patients with purulent meningoencephalitis revealed endothelial cells and infiltrating leukocytes with an increased staining signal for MMP-9, TIMP-1, and TIMP-2 (234). CSF levels of MMP-9 were significantly higher in those children who developed neurologic sequelae than in those who recovered fully, thus identifying high CSF concentrations of MMP-9 as a risk factor for the development of neuronal damage (235).
Studies in a rat model of pneumococcal meningitis documented a 100-fold to 1,000-fold transcriptional induction of MMP-3, -8, -9, -12, -13, and -14, but not of MMP-2 and -7, in brain parenchymal tissue (198,236). In CSF cells, messenger RNA (mRNA) of MMP-8 and -9 were 10-fold to 100-fold increased, whereas MMP-2 and -7 remained at basal levels (198). The cortical brain damage in this experimental model is associated with changes in MMP-9/TIMP-1 ratio and an increase in collagen degradation (237). In experimental meningitis, treatment with MMP inhibitors led to a significant reduction of mortality and seizure incidence and reduced the extent of cortical damage (236,238). In addition to this effect, combined inhibition of MMP and TACE led to a reduction of hippocampal apoptosis and preserved learning capacity of animals that recovered from bacterial meningitis (198).
Neutrophil Invasion
Neutrophil migration into the CSF is a distinctive feature of bacterial meningitis and contributes to the deleterious effects of inflammation on the brain. In response to cytokines, chemokines, and other chemotactic stimuli, neutrophils penetrate the microvascular basement membrane, leaving the bloodstream to accumulate in the CSF, where they produce the profound CSF pleocytosis characteristic of bacterial meningitis (239–241).
The entry of blood-derived neutrophils into the CSF is facilitated by a cascade of events (Fig. 23.2). The initial step in the adhesion cascade is the tethering and rolling of leukocytes along the endothelium, which is mainly mediated by the interaction of members of the selectin family and their glycoprotein ligands (242,243). E-selectin and P-selectin, upregulated on the endothelial cell surface in response to proinflammatory stimuli, bind to P-selectin glycoprotein ligand-1 and other glycosylated ligands (243). The rapid formation and breakup of these bonds on the neutrophil surface enable the rolling motion along the vessel wall (244). Further, the hydrodynamic drag forces close to the vessel wall induce the flattening of the rolling neutrophil. This reduces hydrodynamic drag and increases the area where selectin-ligand bonds may be established (245). Consequently, in mice deficient in P-selectin, partial reduction of neutrophil influx was observed after cytokine-mediated induction of sterile meningitis, whereas mice deficient in P-selectin and E-selectin showed almost complete inhibition of neutrophil influx in the same experimental model (246). The polysaccharide fucoidin is a selectin blocker that inhibits leukocyte rolling and subsequent leukocyte transendothelial migration. Treatment with fucoidin intravenously has been shown to attenuate the pleocytosis in experimental pneumococcal meningitis (247–249). Rolling of leukocytes along a chemotactic gradient is followed by firm adhesion to the endothelium. Halting of the rolling motion is induced by the activation of integrins on the neutrophil surface by chemokines (e.g., IL-8, C5a) (250). Activated leukocyte β1-and β2-integrin (i.e., LFA-1) demonstrate higher affinity to members of the immunoglobulin-like superfamily on the endothelium, including ICAM-1 and vascular cell adhesion molecule-1 (VCAM-1) (251). After firm adhesion to the vessel wall, neutrophils crawl, mediated by a different β2-integrin (i.e., Mac-1), to a suitable transmigration site (252). Transgenic deletion of ICAM-1 in a mouse model of brain inflammation and trauma led to a significant reduction in the number of granulocytes infiltrating the brain tissue (253). Also, antibody-mediated blocking of integrins or ICAM-1 was shown to decrease leukocyte influx in experimental meningitis models, confirming an important role for these molecules in assisting leukocyte entry into inflamed neural tissue (254–256).
Finally, the transmigration of leukocytes across the BBB may occur by the paracellular or transcellular route (243,257,258). Intracellular signaling induced by the interaction of integrins with their ligands leads to remodeling of endothelial cell actin cytoskeleton and facilitates paracellular or transcellular migration of leukocytes (257).
TARGETS OF DAMAGE IN MENINGITIS
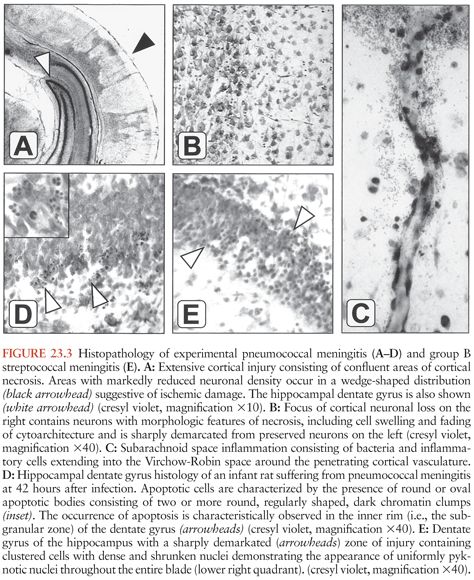
Figure 23.3 shows the histopathology of experimental pneumococcal meningitis and group B streptococcal meningitis.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


