Sensitivity to Complement-mediated Lysis
The chronic intravascular hemolysis that is the clinical hallmark of PNH is due to the abnormal sensitivity of the erythrocytes to complement-mediated lysis. From 1937 to 1939, Ham and Dingle made the seminal observations that connected the hemolysis to complement.
5 Those studies demonstrated that the abnormal cells are hemolyzed when incubated in acidified serum and that the hemolysis is complement-dependent. The lysis of the defective cells in acidified serum (a process that activates the alternative pathway of complement) became the standard technique for the diagnosis of PNH; and appropriately, the assay was called Ham’s test. Approximately 10 years after the studies of Ham and Dingle, cross-transfusion studies confirmed that hemolysis in PNH results from an intrinsic abnormality of the red cell. In those studies, Dacie reported that normal erythrocytes survive normally in patients with PNH, whereas the life span of PNH erythrocytes is shortened both in the patient and in a normal recipient. Not all PNH red cells are equally sensitive to complement-mediated lysis, however, and cohorts of relatively long-lived and very short-lived cells can be distinguished in red cell survival studies. Moreover, the proportions of complement-sensitive and complement-insensitive cells vary greatly among patients. Generally, the percentage of abnormal cells remains stable in an individual patient, although there are clearly exceptions. The percentage of markedly complement-sensitive cells correlates with the clinical course of each patient with respect to the hemolytic component of the disease and perhaps to thrombotic propensity.
A defining feature of PNH is
phenotypic mosaicism based on sensitivity of the erythrocytes to complement-mediated lysis. This remarkable characteristic was first clearly elucidated by Rosse and Dacie in 1966,
6 and Rosse further refined the analysis in 1973.
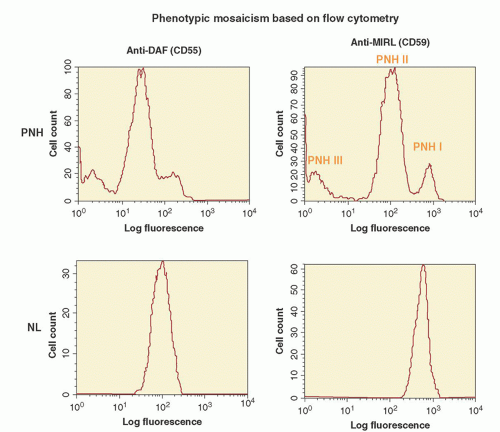
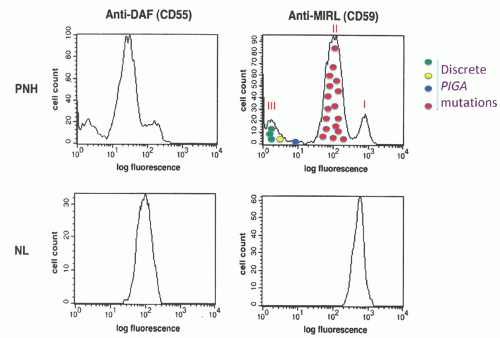
7 Using an in vitro test that quantitates the sensitivity of erythrocytes to complement-mediated lysis (the complement lysis sensitivity assay), three phenotypes of PNH erythrocytes were identified (
Table 31.1;
Figs. 31.1 and
31.2). One of the phenotypes (designated PNH I) was characterized by normal or nearnormal sensitivity to complement, whereas another phenotype (designated PNH III) was 15 to 25 times more susceptible to lysis. A third phenotype (PNH II) was of intermediate sensitivity, about 3 to 5 times more susceptible than normal cells. Most patients have a mixture of type I and type III cells, but mosaics of type I, type II, and type III, as well as type I and type II, are also observed.
As noted above, the proportion of complement-sensitive and -insensitive cells varies greatly among patients. For example, the
erythrocytes of one patient (hypothetical patient A) may be comprised of 10% PNH III cells and 90% PNH I cells, whereas another patient (hypothetical patient B) may have 75% PNH III cells and 25% PNH I cells. The intensity of the hemolytic component of the disease is related to the size of the PNH III population.
8 The proportions of sensitive and insensitive cells usually remain stable for long periods (years to decades); but population shifts may be observed during the course of the disease, and in some patients, the abnormal clone remits spontaneously.
9,
10 The threshold for biochemical evidence of intravascular hemolysis is crossed when the population of GPI-deficient PMNs (as a measure of the PNH clone size) is 20% to 25% or greater.
11 Patients with a clone size of 20% to 25% usually have 3% to 5% PNH erythrocytes (the population of PNH III cells is smaller than the clone size because the complement-sensitive red cells are selectively destroyed intravascularly). As a rule, visible hemoglobinuria is absent when PNH III erythrocytes constitute less than 20% of the red cell population (corresponding to a PNH clone size of 40% to 60%).
8 Paroxysms of gross hemoglobinuria occur when the PNH III population ranges from 20% to 50% of the population (corresponding to a PNH clone size of 60% to 90%), and constant hemoglobinuria is associated with greater than 50% PNH III erythrocytes (PNH clone
size >90%). PNH II cells (the erythrocytes of intermediate sensitivity to complement), even when present in high proportions, are associated with no visible hemoglobinuria.
The proportion of abnormal cells is greater in the marrow than in the blood and, among circulating erythrocytes, is greatest in young cell populations (the percentage of GPI-deficient reticulocytes is similar to the percentage of GPI-deficient PMNs).
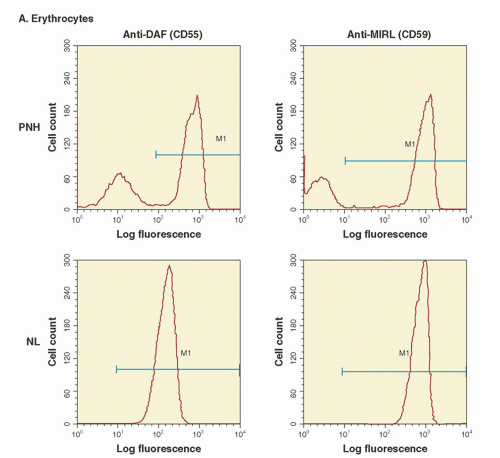
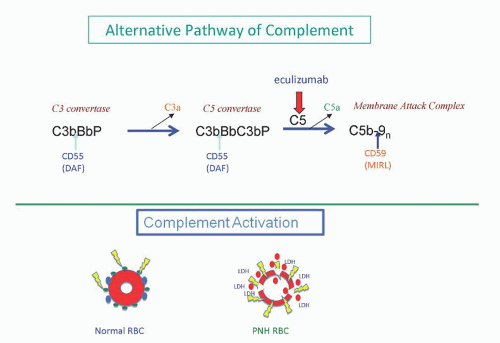
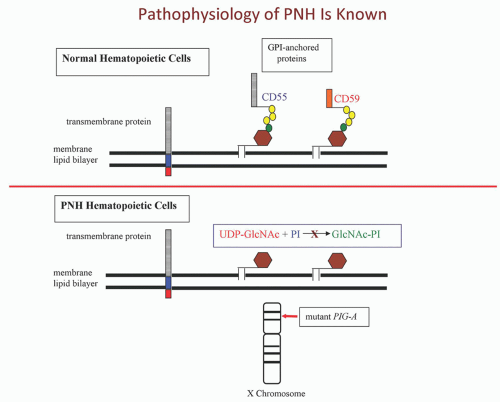
PNH is not an immune-mediated hemolytic anemia that arises as a consequence of the development of autoantibodies. Rather, the failure to regulate an alternative pathway of complement activation on the erythrocyte surface underlies the hemolysis of PNH (
Fig. 31.3). Under physiologic conditions, the alternative pathway is in a state of continuous, low-grade activation. Normal erythrocytes are not hemolyzed as a result of this low-grade alternative pathway activation because specific cell-surface proteins have evolved that inhibit the activity of complement (
Fig. 31.3). In contrast, PNH erythrocytes are deficient in the two most important membrane regulators of complement (CD55 and CD59), and consequently, they are subject to chronic, spontaneous hemolysis in vivo (
Fig. 31.3). Failure to inhibit alternative pathway activation has a profound effect on red cell survival, as studies have shown that the half-life of complement-sensitive PNH cells is approximately 6 days (compared to 60 days for normal erythrocytes).
Rosse and colleagues reported the first clear evidence of the nature of the aberrant interactions of PNH erythrocytes with complement in 1973 and 1974.
12,
13 Those investigators showed that when complement is activated in vitro by either the classical or the alternative pathway, PNH erythrocytes bind much greater amounts of activated C3 than normal erythrocytes. The difference in C3 deposition was particularly striking when acidified serum was used to activate the alternative pathway. Subsequent studies showed that PNH red cells lacked the capacity to regulate the formation and stability of the amplification C3 convertases of complement,
14,
15 thus accounting for the greater binding of activated C3. A second defect was suggested by experiments demonstrating that PNH erythrocytes also failed to regulate the activity of the cytolytic membrane attack complex of complement.
16,
17 Collectively these observations indicated that PNH erythrocytes had two complement regulatory defects, one that affected regulation of the C3 convertases and a second that affected regulation of the membrane attack complex (
Fig. 31.3).
Molecular Basis of Paroxysmal Nocturnal Hemoglobinuria
The observation that, based on sensitivity to complement, the peripheral blood of PNH patients is a mosaic comprised of both normal and abnormal cells suggested that the abnormal cells were the progeny of a mutant clone and that they coexisted with the progeny of residual normal stem cells. Further, that PNH is an acquired rather than an inherited disease implied that the abnormal clone arises as a consequence of a somatic mutation. In 1970, Oni et al.
41 analyzed G6PD isoforms in both the complement-sensitive and the complement-insensitive erythrocytes of a female PNH patient who was heterozygous at the G6PD locus.
The complement-insensitive cells expressed both G6PD isoforms, indicating a polyclonal origin for this cohort. In contrast, the complement-sensitive cells expressed only one isoform, a finding consistent with monoclonality. These studies provided the first experimental evidence in support of the clonal hypothesis of PNH. In 1969, Aster and Enright
42 reported that a portion of both the platelet and granulocyte populations from patients with PNH was abnormally sensitive to complement-mediated cytolysis. This publication represented another watershed event in the understanding of the origins of PNH, because it indicated that the mutation arose in a primitive hematopoietic stem cell that has the capacity to differentiate along myeloid lines. Subsequent studies showed that monocytes are also affected, and, in most patients, affected lymphocytes were demonstrated. Together with the observation that all proteins that are deficient in PNH are GPI-anchored, these studies suggested that PNH arises as a result of a somatic mutation affecting a pluripotent hematopoietic stem cell and that the gene that is mutant is essential for the normal biosynthesis of the GPI anchor.
The GPI anchor is a complex structure with at least 25 proteins being essential for assembly of the moiety.
43,
44 Hypothetically, the PNH phenotype would result if any of these proteins were nonfunctional, because if the GPI anchor is not synthesized, GPI-anchored proteins are not expressed. Accordingly, it seemed probable that PNH would be found to be heterogeneous at the molecular level as mutations affecting any one of several genes that encode elements critical for GPI anchor assembly would produce the disease phenotype. In 1992
45 and 1993,
46 however, two groups published the surprising finding that GPI-protein deficient lymphocyte cell lines derived from different patients with PNH all belonged to the same complementation class (Class A) of GPI-AP-deficient cell lines. Additional experiments confirmed that the PNH cell lines had the same biochemical defect as the complementation Class A mutants. Like the Class A mutants, the PNH cell lines failed to synthesize N-acetylglucosaminyl-phosphatidylinositol (GlcNAc-PI), the first intermediate in the pathway of GPI anchor assembly (
Fig. 31.4).
A gene that restores normal expression of GPI-anchored proteins in the complementation Class A PNH cell lines was identified by Kinoshita and colleagues in 1993.
47,
48 As predicted by the studies cited above, the gene, called
PIGA, encodes a protein that is essential for the normal synthesis of GlcNAc-PI.
47 Subsequently, Takeda et al.
48 showed that
PIGA complements the deficient expression of GPI-anchored proteins in PNH lymphoblastoid cell lines and that those cell lines harbored somatic mutations in
PIGA. Together, those studies defined both the biochemical and the molecular basis of the deficiency of GPI-anchored proteins in PNH (
Fig. 31.4).
Additionally, Takeda and colleagues observed that a heterozygous mutation was sufficient to produce the PNH phenotype.
48 As presaged by the studies of Hyman et al.,
49 the dominant expression of the somatic mutation was explained when
PIGA was mapped to chromosome Xp22.1. As males have a single X chromosome, any functionally significant mutation affecting
PIGA is expressed. Females are functionally haploid due to X-inactivation. Therefore, somatic mutations in
PIGA appear dominant when they occur on the active X chromosome. To date, somatic mutations affecting
PIGA account for the PNH phenotype in all patients in whom the genetic basis has been identified.
PIGA mutations have not been identified in all patients studied, but this apparent discrepancy is likely a technical artifact rather than a true difference in etiology. The chromosomal location of
PIGA provides the most logical explanation for the uniformity of the molecular defect in PNH. This interpretation supposes that all other genes that are essential for synthesis of the GPI moiety are autosomal, a supposition supported by available data.
Analysis of
PIGA has revealed that the same mutation can be identified in isolated neutrophils, monocytes, and lymphocytes from individual patients with PNH, confirming that the disease involves the hematopoietic stem cell.
48,
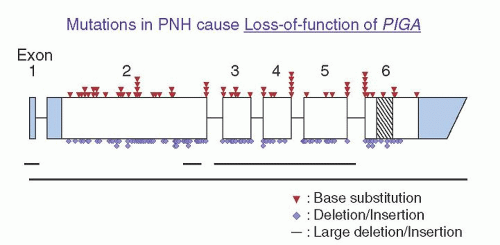
50 More than 150
PIGA mutations have been identified in affected cells of patients with PNH. Only three large deletions have been observed
51,
52 (
Fig. 31.5). The remaining mutations consist of nucleotide substitutions of the missense or nonsense types, or small deletions or insertions that cause frameshifts and introduce premature termination codons (
Fig. 31.5). The mutations are distributed randomly over the entire coding region and at splice junctions (
Fig. 31.5). At least 19 mutations have been observed in more than one patient. Absence of repetitive mutations indicates that
PIGA lacks molecular hot spots. The simplest explanation for these observations is that clonal selection and expansion depends on complete or nearly complete inactivation of PIGA such that expression of GPI-AP proteins is below a critical threshold.
Therefore all PIGA mutations in PNH are loss-of-function mutations.Studies of
PIGA mutations have also provided insights into the molecular basis of the phenotypic mosaicism of PNH.
50 Cloned lymphocyte cell lines were established from the peripheral blood of a patient whose erythrocytes were a mixture of PNH I, PNH II, and PNH III cells (
Fig. 31.1). Based on expression of GPI-anchored proteins, lymphocyte clones with four different phenotypes were observed. Analysis of clones with normal expression of GPI-AP revealed no somatic
PIGA mutations. In contrast, among the three phenotypically distinct lymphocyte clones with abnormal GPI-AP expression, four discrete
PIGA mutations were identified. In the lymphocyte clones with the PNH II phenotype, a missense mutation that changed a highly conserved amino acid was found. This finding suggests that cells with partial expression of GPI-anchored proteins (PNH II) are derived from stem cells with mutations that
incompletely inactivate PIGA. In the case of the lymphocyte clones with the PNH III phenotype, three separate mutations were identified, each of which was expected to inactivate completely the
PIGA gene product. Collectively, these experiments demonstrate that the phenotypic mosaicism that is characteristic of PNH is a consequence of genotypic mosaicism. Further, because any mutation that completely inactivates PIGA results in PNH III cells, phenotypically identical cells can have different
PIGA genotypes (
Fig. 31.6).
Studies of the pattern of X chromosomal inactivation indicated that, in the female patient with four different
PIGA mutations, the abnormal clones were not derived from a common ancestor.
50 Further, that each of the mutations was discrete demonstrated that the mutational events occurred independently rather than by clonal evolution. These results demonstrate that PNH is not strictly a monoclonal process and that they have important implications for the origins of the disease.
Studies of mutational frequency in PNH have produced conflicting results. Some of the disparity is almost certainly due to differences in experimental design and interpretation of data. While an abnormally high mutational rate may contribute to generation of multiple PIGA-mutant clones, a selective advantage for GPI-AP-deficient stem cells and variable extent of expansion of the mutant clones appears to be necessary to explain the outgrowth of multiple PNH clones, some of which dominate hematopoiesis and some of which persist subclinically.
The Pathophysiology of Paroxysmal Nocturnal Hemoglobinuria Is Unique
Despite the progress that has been made in determining the basis of the abnormal sensitivity of the erythrocytes to complement-mediated lysis and the global absence of GPI-anchored proteins (GPI-AP) from hematopoietic cells, an issue that is fundamental to a more complete understanding of PNH remains largely enigmatic. In order for PNH to become clinically evident, the hematopoietic
stem cells bearing the mutant
PIGA must expand so that progeny sufficient to produce symptoms and signs of the disease are generated. In many instances, GPI-AP-deficient (GPI-AP
−) cells dominate hematopoiesis in patients with PNH, suggesting that the mutant stem cell has either a greater proliferative capacity or a survival advantage relative to GPI-AP sufficient (GPI-AP
+) stem cells. That
PIGA mutations are necessary for the development of PNH is incontrovertible. At issue is whether
PIGA mutations are both necessary
and sufficient to account for the PNH syndrome and whether the
PIGA mutation provides an absolute or a conditional growth/survival advantage.
PNH differs from monoclonal hematopoietic stem cell disorders such as chronic myelogenous leukemia in which the t(9;21) that generates the fusion protein BCR-ABL is sufficient to account for the proliferative advantage of the mutant cell, and in which all normal hematopoiesis is progressively and invariably displaced as a consequence of the uncontrolled proliferation of a transformed clone. In PNH, the peripheral blood is a mosaic of normal and abnormal cells and the proportion of GPI-AP−:GPI-AP+ cells varies greatly among patients; that ratio tends to remain fixed over long periods of observations. PNH is, therefore, more aptly characterized as having deregulated cellular proliferation and expansion, rather than cellular transformation.
The oligoclonal nature of PNH
50 suggests that a powerful selection process that is most likely based on phenotype is at work in the bone marrow. According to this hypothesis, stem cells with mutant
PIGA have an advantage because of some pathological process (perhaps immune-mediated) that involves a GPI-AP. For example, an autoimmune process could arise in which the target antigen is a GPI-AP expressed on hematopoietic stem cells. Under those circumstances,
PIGA-mutant stem cells (lacking GPI-AP) would escape immune-mediated destruction because the target antigen is absent.
Hematopoietic cells express a relatively large number of functionally diverse GPI-AP (
Table 31.2). Thus, absence of all GPI-AP is probably not required for the clonal selection of the mutant stem cells. Rather, the selective advantage may be dependent on the absence of a single protein that is GPI-anchored, and the reason for the global deficiency of GPI-AP is that
PIGA is located on the X chromosome. According to this supposition, an autosomal gene encodes the GPI-AP that is conditionally detrimental (e.g., an antigen targeted for immune destruction or a receptor for a negative growth regulator). Inasmuch as two alleles rather than one must be mutated, the probability of inactivating an autosomal gene through somatic mutagenesis is remote compared with the probability of inactivating an X-linked gene. Therefore, stem cells with a deficiency of the detrimental GPI-AP are most likely to arise as a consequence of
PIGA mutations, as is the case in patients with CLL treated with Campath1H (anti-CD52).
53 Assuming that the GPI-AP complement regulatory proteins are not the targets of the underlying pathologic process, the hemolytic anemia that is the clinical hallmark of PNH may represent an epiphenomenon related to the chromosomal location of
PIGA.
Although a compelling case can be made in support of the paradigm that the
PIGA mutation bestows a conditional growth advantage upon the affected stem cell, the hypothesis that the mutant stem cells have an absolute growth or survival advantage must also be considered. Data that both support and challenge this hypothesis can be found.
54,
55,
56 The most compelling argument against an intrinsic growth or survival advantage for
PIGA-mutant cells is made by the results of studies using transgenic mice.
57 By using homologous recombination, Kawagoe et al.
58 disrupted
Piga (the murine homologue of
PIGA). Only mice with a low degree of chimerism survived. Among those animals, the percentage of GPI-AP
− erythrocytes ranged from ˜1% to 5%. During 10 months of observation, the ratio of GPI-AP
−: GPI-AP
+ peripheral blood cells did not increase. Studies by others using conditional knockout technology have confirmed these observations.
57
A Two-Step Model of Paroxysmal Nocturnal Hemoglobinuria Pathogenesis
Studies by Inoue and colleagues suggest that a two-step mechanism may account for the unique pathophysiology of PNH.
61 Those investigators identified two patients whose
PIGA-mutant cells had a concurrent, acquired rearrangement of chromosome 12. Detailed analysis showed that in both cases, the chromosome 12 rearrangement resulted in disruption of the 3′ untranslated region of
HMGA2. As a consequence, a negative regulatory region of the locus was disrupted, resulting in ectopic expression of the gene.
HMGA2 is a member of the high mobility group of proteins (
HMGA1a,
HMGA1b,
HMGA2) that function as architectural transcription factors. HMG members possess no intrinsic transcriptional activity. Instead, these nonhistone phosphoproteins orchestrate assembly of stereospecific transcriptional regulatory proteins into enhanceosomes. The cellular targets of
HMGA2 are incompletely defined but appear to include cyclin A and E2F1.
Molecular studies established a causal role for
HMGA2 in benign mesenchymal tumors. Rearrangement of 12q13-15 is observed in these neoplasms, but tumorigenesis does not depend on generation of chimeric proteins derived from fusion of
HMGA2 with specific translocation partners. Rather, clonal expansion induced by
HMGA2 appears to result from deregulated expression of the protein. For the two PNH patients, ectopic expression was a consequence of gain-of-function mutational events caused by disruption of the 3′UTR shown to contain elements that negatively regulate
HMGA2 mRNA stability, including the microRNA let-7a.
62,
63 Additional studies will be required to determine whether aberrant expression of
HMGA2 underlies clonal expansion in PNH patients without structural abnormalities of 12q13-15, but studies by Murakami and colleagues support this hypothesis.
64In contrast to a disorder characterized by cellular transformation and malignant cellular growth, PNH manifests many of the characteristics of a benign tumor, since there is limited expansion of
PIGA-mutant clones (the peripheral blood of patients is a relatively stable mosaic of normal and abnormal cells);
PIGA-mutant cells respect tissue boundaries (there is no invasion of non-hematopoietic tissues);
PIGA-mutant cells respond appropriately to signals that normally regulate hematopoiesis (function is not autonomous); and transformation into acute leukemia occurs rarely (PNH is not a premalignant condition).
65 The studies of Inoue and colleagues
61 suggest the concept of PNH as a benign tumor of the bone marrow with aberrant expression of
HMGA2 acting in concert with mutant
PIGA (and the consequent deficiency of GPI-APs) to produce the proliferative phenotype that underlies clonal expansion. These studies did not define how the aberrant expression of
HMGA2 works additively or synergistically with mutant
PIGA to produce the proliferative phenotype.
The findings of Inoue et al. provide new insights into the etiology of the nonmalignant clonal hematopoiesis of PNH. These studies support a two-step process consisting of (i) clonal selection based on phenotype (i.e., GPI-AP deficiency resulting from mutant PIGA) and (ii) clonal expansion as a consequence of a second somatic mutation that bestows an additional growth/survival advantage. Clonal selection may induce the exit of PIGA-mutant stem cells from a dormant state, thereby favoring acquisition of the second mutation that underlies clonal expansion. The variable expansion of PIGA-mutant clones among patients and among mutant clones in the same patient could be explained by heterogeneity involving the second genetic event that partners with mutant PIGA. According to this hypothesis, mutant PIGA is promiscuous with respect to the second mutant gene with which it partners. But the benign nature of PNH suggests that genes involved in clonal expansion of PIGA-mutant stem cells are different from those that underlie malignant clonal diseases such as acute leukemia.
In summary, the basis of the clonal dominance in PNH is incompletely understood; however, available evidence supports a two-step model of pathogenesis. Step 1 of this model is
clonal selection. In this case, a loss-of-function mutation affecting
PIGA causes deficiency of GPI-APs on affected stem cells. These mutant stem cells have a relative growth or survival advantage that is likely enhanced in the setting of bone marrow failure (e.g., aplastic
anemia). Thus, principles of Darwinian evolution may apply to development of PNH.
Clonal expansion, step 2 of the PNH pathogenesis model, is envisioned as a consequence of clonal evolution in which a second somatic (gain-of-function) mutation working in concert with the consequences of mutant
PIGA bestows upon the double mutant stem cell an absolute proliferative advantage.
61 The extent of expansion of a particular mutant clone and the dominance of one
PIGA-mutant clone in the presence of other
PIGA mutants may be mediated by the nature of the second event that may be molecularly heterogeneous. Alternatively, a limited number of second mutations may partner with mutant PIGA, and the extent of clonal expansion may be determined by how the second mutation affects gene function.
Aplastic Anemia and Paroxysmal Nocturnal Hemoglobinuria
Patients with
clinical PNH can be divided into the following two groups: those without a preceding history of aplastic anemia (classic PNH); and those with an antecedent history of aplastic anemia who subsequently develop PNH (PNH/aplastic anemia).
1 An association between aplastic anemia and PNH has been recognized at least since 1961, and numerous subsequent studies have confirmed the association. The probability is negligible that these two rare diseases would occur together so frequently by chance. Therefore, a pathophysiologic link between PNH and aplastic anemia must exist.
Hillmen et al.
65 reported that 23 of 80 PNH patients (29%) had an antecedent history of aplastic anemia, and in a series of 220 French patients with PNH, Socié and colleagues identified 65 (30%) in whom the diagnosis of aplastic anemia preceded that of PNH.
66 The latter group of investigators also found that 10% of PNH patients with no antecedent history of aplastic anemia developed pancytopenia during the period of observation covered by the study (median follow-up of 2 years). The estimated cumulative incidence of pancytopenia was 8.2% (± 2.4%, SE) at 2 years and 14.2% (± 3.3%, SE) at 4 years.
66 This incidence appears to be somewhat higher than that observed in the study reported by Hillmen et al. in which 5 of 80 patients (6%) with PNH subsequently developed aplasia.
65On the other hand, the proportion of patients with aplastic anemia who subsequently develop PNH varies widely among studies, in part because the criteria for diagnosis of PNH are not uniform. In some cases, a positive Ham’s test or sucrose lysis test was required for diagnosis, and in other cases, identification by flow cytometry of a population of peripheral blood cells with GPI-AP deficiency was used to classify patients. A recent study reported that PNH cells were found in the BM in 89% of 115 patients with aplastic anemia at some point during the course of their illness.
67 Two other studies reported 38% (11 of 29 patients)
68 and 32% (12 of 37 patients)
69 for the portion of patients with aplastic anemia treated with immunosuppression who developed laboratory evidence of PNH during the course of their disease. In the latter study, 23% of patients had clinical signs and symptoms of PNH. Therefore, in most patients with aplastic anemia, evidence of PNH is subclinical or transient. These results suggest that the selective pressure that favors
PIGA-mutant cells occurs frequently in the setting of aplastic anemia, but that the clinical syndrome occurs at a significantly lower frequency.
The time between the diagnosis of aplastic anemia and the development of PNH varies from a few months to several years. In the series reported by Socié et al.,
66 the median time between diagnosis of aplastic anemia and laboratory evidence of PNH was 3.1 years (range 0.17 to 15 years). Using an assay with the capacity to detect 0.004% GPI-AP-deficient erythrocytes, Mukhina et al. reported that 61% of patients with aplastic anemia had detectable PNH cells prior to therapy.
70 Similar results were obtained in more recent studies using high-resolution flow cytometry.
71 In most of these cases, the GPI-AP-deficient cells would be undetected by conventional flow cytometry, in which the sensitivity threshold for detecting cells with the PNH phenotype is 1% to 3%. Identification of very small populations of GPI-AP-deficient cells by high-resolution flow cytometry has led to a new class of PNH, termed
subclinical PNH (PNH-sc).
1 Patients with PNH-sc have no clinical or laboratory evidence of hemolysis. Small populations of GPI-AP-deficient hematopoietic cells (peripheral blood erythrocytes, granulocytes, or both) are detected by very sensitive flow cytometric analysis. PNH-sc is observed in association with bone marrow failure syndromes, particularly aplastic anemia and the refractory anemia variant of MDS.
In summary, while 40% to 60% of patients with aplastic anemia have small, subclinical populations of GPI-AP
− hematopoietic cells at diagnosis,
71 only 10% to 15% subsequently develop clinically apparent PNH.
72,
73 In the remainder, GPI-AP
− cells persist subclinically or disappear,
71,
74 suggesting that mutant
PIGA (and the consequent deficiency of GPI-APs) is necessary for clonal selection but is insufficient to account for the clonal expansion required for clinical manifestations of PNH to become apparent. The simplest interpretation of these observations is that factors in addition to mutant
PIGA contribute to the development of clinical signs and symptoms of PNH by affecting the extent to which the
PIGA-mutant stem cells expand.
As expansion of the mutant clones occurs later in the course of aplastic anemia, the process could be influenced directly or indirectly by therapy. Although many patients with aplastic anemia who develop PNH are treated with immunosuppressive therapy (e.g., anti-thymocyte globulin and cyclosporin), there is no evidence that immunosuppression causes PNH.
73 Patients with aplastic anemia who respond to androgens appear equally likely to develop PNH.
66The basis of the relationship between PNH and aplastic anemia is speculative. Essentially all patients with PNH have some evidence of bone marrow failure (e.g., thrombocytopenia, leukopenia, or both) during the course of their disease. Therefore, bone marrow injury may play a central role in the development of PNH by providing the conditions that favor the growth/survival of
PIGA-mutant, GPI-AP-deficient stem cells. Currently, there is no evidence that the types of
PIGA mutations that occur in PNH/aplastic anemia are different from those observed in classic PNH.
75 Further, a distinction between classic PNH and PNH/aplastic anemia may be artificial, as the underlying pathophysiologic process could be the same. According to this hypothesis, in classic PNH, the aplastic or hypoplastic component is subclinical and short-lived, with disease noted only after the
PIGA-mutant clone has expanded sufficiently to dominate hematopoiesis.
Leukocytes and Platelets
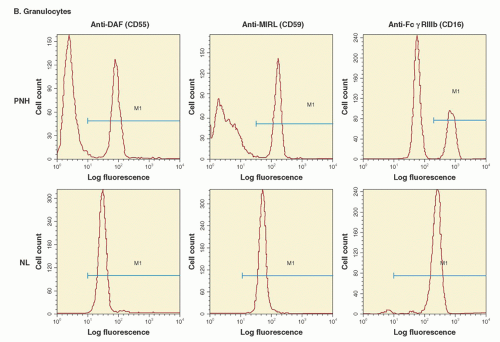
Deficiency of GPI-AP on neutrophils, monocytes, platelets, and lymphocytes has been demonstrated in the peripheral blood of patients with PNH, and identical
PIGA mutations have been identified in neutrophils, monocytes, and lymphocytes from the same patient.
48,
50 Together, these studies indicate that the somatic mutation that gives rise to PNH affects a hematopoietic stem cell. Most PNH patients have pancytopenia or either neutropenia or thrombocytopenia in combination with anemia at some point during the course of their illness. The neutropenia and thrombocytopenia, however, are due to abnormal hematopoiesis rather than to increased peripheral, complement-mediated destruction, as in vivo studies have demonstrated normal survival of neutrophils and platelets in patients with PNH. That absence of GPI-anchored complement regulatory proteins from PNH neutrophils and platelets does not affect their survival implies that these cell types (unlike erythrocytes) have mechanisms in addition to those provided by CD55 and CD59 that protect them from complement-mediated destruction in vivo.
In vitro studies have shown functional abnormalities of PNH leukocytes and platelets. Further, deficiency of some of the GPI-AP from PNH leukocytes and platelets would seem to have important functional consequences (e.g., deficiency of Fc
γRIIIb from neutrophils, deficiency of urokinase-type plasminogen activator receptor from monocytes and neutrophils, deficiency of LFA-3 from lymphocytes, or deficiency of the folate receptor from hematopoietic stem cells) (
Table 31.2). However, evidence that deficiency of GPI-APs other than erythrocyte CD55 and CD59 contributes to the pathophysiologic manifestations of PNH is largely conjectural. At least in some cases, functional redundancy appears to account for the lack of untoward consequences associated with GPI-AP deficiency.
 The Diagnostic and Therapeutic Approach to Hematologic Problems
The Diagnostic and Therapeutic Approach to Hematologic Problems



