INTRODUCTION
SUMMARY
In contrast to all other intrinsic abnormalities of the erythrocyte, paroxysmal nocturnal hemoglobinuria (PNH) is an acquired, not an inherited, disorder. PNH arises as a consequence of somatic mutation, involving one or more hematopoietic stem cells, of PIGA, a gene located on the X chromosome that is required for synthesis of the glycosylphosphatidylinositol (GPI) moiety that anchors some proteins to the cell surface. Consequently, all GPI-anchored proteins (GPI-APs) that are normally expressed are deficient on the mutant hematopoietic stem cells and their progeny. The complement-mediated intravascular hemolytic anemia and the resulting hemoglobinuria that are the clinical hallmarks of PNH are a consequence of deficiency of the GPI-anchored complement regulatory proteins, CD55 and CD59. Although PNH is a neoplastic (clonal) disease, it is not a malignant disease in that there is no exaggerated proliferation of neoplastic cells and replacement of marrow or spread to other tissues, and the extent to which the mutant clones expand varies greatly among patients. Thus, the blood cells of patients with PNH are a mosaic of phenotypically normal and abnormal cells. The size of the mutant clone is an important determinant of the clinical manifestations of the disease, which include hemolysis, thrombophilia, and, in many patients, pancytopenia as a result of marrow failure. The diagnosis of PNH is confirmed using flow cytometry to detect and quantify the percentage of blood erythrocytes and leukocytes (i. e., neutrophils and monocytes) that lack GPI-APs measured as intensity of CD55 and CD59 on the cell surface. The intravascular hemolysis of PNH can be controlled with eculizumab, a humanized monoclonal antibody that blocks formation of the cytolytic membrane attack complex of complement. Although treatment with eculizumab favorably modifies the natural history of PNH, it has no effect on the underlying disease process (i.e., the PIGA-mutant hematopoietic stem cell clone). The PIGA-mutant mutant clone can be eradicated and normal hematopoiesis restored by allogeneic hematopoietic stem cell transplantation, but the relatively benign natural history of PNH in patients treated with eculizumab has tempered enthusiasm for transplantation because of concerns about subjecting patients to the risk of treatment-related morbidity.
Acronyms and Abbreviations:
APC, alternative pathway of complement; CD55, an antigen encoding DAF; CD59, an antigen encoding MAC-inhibitory protein; DAF, decay-accelerating factor; GPI, glycosylphosphatidylinositol; GPI-APs, glycosylphosphatidylinositol-anchored proteins; GVHD, graft-versus-host disease; HLA, human leukocyte antigen; INR, international normalized ratio of prothrombin assay data; LDH, lactate dehydrogenase; MAC, membrane attack complex of complement; MDS, myelodysplastic syndrome; MIRL, membrane inhibitor of reactive lysis; PIGA, phosphatidylinositol glycan class A; PMN, polymorphonuclear cell; PNH, paroxysmal nocturnal hemoglobinuria; PNH-sc, subclinical PNH; RA, refractory anemia; RAEB, refractory anemia with excess of blasts; RAEB-t, refractory anemia with excess of blasts in transformation; RA-PNH+, RA with a population of PNH cells; RA-PNH−, RA without a population of PNH cells; RARS, refractory anemia with ringed sideroblasts; RBCs, red blood cells; RCMD, refractory cytopenias with multilineage dysplasia; WHO, World Health Organization.
DEFINITION AND HISTORY
Although commonly regarded as a type of hemolytic anemia, paroxysmal nocturnal hemoglobinuria (PNH) is properly categorized as a hematopoietic stem cell disorder. PNH arises as a result of clonal expansion of one or several hematopoietic stem cells that have acquired a somatic mutation of the X-chromosome gene PIGA (phosphatidylinositol glycan class A). As a consequence of mutant PIGA, any progeny of affected stem cells (erythrocytes, granulocytes, monocytes, platelets, and lymphocytes) are deficient in all glycosylphosphatidylinositol-anchored proteins (GPI-APs) that are normally expressed on hematopoietic cells. The clinical manifestations of PNH are hemolytic anemia, thrombophilia, and marrow failure, but only the hemolytic anemia is unequivocally a consequence of somatic mutation of PIGA. It is not a malignant neoplasm in the classical sense of uncontrolled proliferation of cells, spread to tissues other than marrow, or spatial replacement of hematopoiesis. Its effects can be lethal and it can uncommonly undergo clonal evolution to acute myelogenous leukemia.
Comprehensive, scholarly reviews of the history of PNH have been published.1,2,3,4 The first published clinical description of PNH is attributed to William Gull in 1866, but he failed to distinguish definitively PNH from paroxysmal cold hemoglobinuria. Paul Strübing, in 1882, clearly recognized PNH as a distinct entity and undertook prescient experiments designed to test his hypothesis that the nocturnal hemoglobinuria was a consequence of acidification of plasma that occurred when carbon dioxide and lactic acid accumulated because of slowing of respiration during sleep. In 1911, A.A. Hijmans van den Berg demonstrated that the hemolysis of PNH is caused by a defect in the red cell rather than by the presence of an abnormal plasma factor (as is the case with paroxysmal cold hemoglobinuria; Chap. 54). Thomas Hale Ham is credited with discovering, in the late 1930s, that complement mediates the hemolysis of PNH erythrocytes, although it was not until the alternative pathway of complement was identified and characterized in the mid-1950s by Louis Pillemer that the basis of Ham’s original observations became apparent. Ham developed the acidified serum lysis test (Ham test) that, along with the sucrose lysis test (sugar water test) of Robert Hartmann and David Jenkins, was used as the standard diagnostic test for PNH until being supplanted in the early 1990s by flow cytometry. Both Hartmann and William Crosby brought attention to the important role that thrombosis (particularly the Budd-Chiari syndrome) plays in the natural history of PNH, and John Dacie and his student and colleague S.M. Lewis were the first to systematically characterize the relationship between PNH and marrow failure.
EPIDEMIOLOGY
The prevalence of PNH is not known with certainty. Prevalence estimates are influenced by bias in study design and results differ considerably, in large part, because of the heterogeneous nature of the disease. The blood of patients with PNH is a mosaic of normal and abnormal cells, and the extent of the mosaicism varies widely among patients (see “Phenotypic Mosaicism is Characteristic of Paroxysmal Nocturnal Hemoglobinuria” below). Patients with small PNH clones have few or no symptoms related to hemolysis. Thus an argument can be made that asymptomatic patients with small clones do not have clinically significant PNH and should be excluded from prevalence estimates. Others, however, may argue that any patient with flow cytometric evidence of a population of GPI-AP–deficient cells, regardless of clone size, has PNH and should be included in prevalence estimates. Well-designed, rigorous studies of prevalence that address the issue of disease heterogeneity are needed, but, by any definition, PNH is a rare disease. The prevalence of clinically significant PNH (i.e., classic PNH) plus patients with relatively large clones that arise in the setting of another marrow failure syndrome, (see “Clinical Features” and Table 40–2 below) is likely in the order of less than 1 case per 200,000 population, easily fulfilling criteria (<1 case per 50,000 population) for classification as an ultraorphan disease.5 There is a close association between PNH and aplastic anemia, and environmental factors, drugs, and toxins, that cause aplastic anemia, concordantly increase the risk of developing PNH. Although PNH is reported in all age groups, the peak incidence is in the third and fourth decades of life, similar to that of aplastic anemia (Chap. 35). PNH is an acquired disorder, and there is no known inherited risk for developing the disease. A number of cases have been reported in which only one of a pair of identical twins was affected.
ETIOLOGY AND PATHOGENESIS
The chronic intravascular hemolysis that is the hallmark clinical manifestation of PNH is mediated by the alternative pathway of complement (APC) (Fig. 40–1).6 The APC is a component of innate immunity (Chap. 20).7 This ancient system evolved to protect the host against invasion by pathogenic microorganisms. Unlike the classical pathway of complement that is part of the system of acquired immunity and requires antibody for initiation of activation, the APC is in a state of continuous activation, armed at all times to protect the host (Chap. 19 provides a detailed review of the complement system). The APC cascade can be divided into two functional components: the amplification C3 and C5 convertases and the membrane attack complex (MAC). The C3 and C5 convertases (Fig. 40–1, top panel) are enzymatic complexes that initiate and amplify the activity of the APC. Generation of C5b by enzymatic cleavage of C5 by the APC C5 convertase activates the terminal pathway of complement that results ultimately in assembly of the cytolytic MAC.
Figure 40–1.
Complement-mediated lysis of paroxysmal nocturnal hemoglobinuria (PNH) erythrocytes. Upper panel. The hemolytic anemia of PNH is Coombs test–negative because the process is mediated by the antibody-independent alternative pathway of complement (APC). The C3 convertase of the APC consists of activated C3 (C3b), activated factor B (Bb, the enzymatic subunit of the complex), and factor P (a protein that stabilizes the complex, formally called properdin). The C5 convertase has the same components as the C3 convertase except that two C3b molecules are required to bind and position C5 for cleavage by activated factor B (Bb). C3a and C5a are bioactive peptides that are generated by cleavage of C3 and C5, respectively, by their specific activation convertases. The C3 and C5 convertases greatly amplify complement activation by cleaving multiple substrate molecules. The membrane attack complex (MAC) consists of activated C5 (C5b), C6, C7, C8, and multiple molecules of C9 (C9n). The MAC is the cytolytic unit of the complement system. The glycosylphosphatidylinositol (GPI)-anchored complement regulatory protein CD55 restricts formation and stability of both the C3 and the C5 amplification convertases by destabilizing the interaction between activated factor B (Bb) and C3b (indicated by the blue arrow), whereas GPI-anchored CD59 blocks formation of the MAC by inhibiting the binding of C9 to the C5b-8 complex (indicated by the brown arrow). Inhibition of MAC formation by the humanized monoclonal anti-C5 antibody eculizumab (indicated by the red arrow) ameliorates the intravascular hemolysis of PNH. Lower panel. Normal erythrocytes (left) are protected against complement-mediated lysis primarily by CD55 (blue circles) and CD59 (green circles). Deficiency of these GPI-anchored complement regulatory proteins results in APC activation on PNH erythrocytes (right). Because of deficiency of CD55 and CD59, the complement cascade activates on the cell surface. Consequently, MACs form pores in the red cell membrane resulting in colloid osmotic lysis and release of hemoglobin (red circles) and other contents of the red cell including lactate dehydrogenase (LDH) into the intravascular space. RBC, red blood cell. (Modified with permission from Parker CJ: The pathophysiology of paroxysmal nocturnal hemoglobinuria. Exp Hematol 35(4):523–533, 2007.)
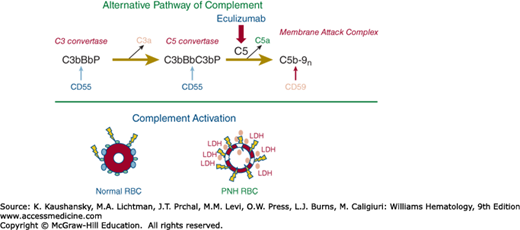
Because the APC is primed for attack at all times, elaborate mechanisms for self-recognition and for protection of the host against APC-mediated injury have evolved. Both fluid-phase and membrane-bound proteins are involved in these processes. Normal human erythrocytes are protected against APC-mediated cytolysis primarily by decay-accelerating factor (DAF, CD55)8,9,10 and membrane inhibitor of reactive lysis (MIRL, CD59).11 These proteins act at different steps in the complement cascade (see Fig. 40–1, top panel). CD55 regulates the formation and stability of the C3 and C5 convertases, whereas CD59 blocks the formation of the MAC. Deficiency of CD55 and CD59 on the erythrocytes of PNH is the pathophysiologic basis of the Coombs-negative, intravascular hemolysis that is the clinical hallmark of the disease (see Fig. 40–1, bottom panel). But why are PNH erythrocytes deficient in the two complement regulatory proteins?
PNH is a consequence of clonal expansion of one or more hematopoietic stem cells with mutant PIGA(located on Xp22.1).12 The protein product of PIGA is a glycosyl transferase12,13,14,15,16 that is an obligate constituent of a complex biochemical pathway required for synthesis of the glycosylphosphatidylinositol (GPI) moiety that anchors individual proteins belonging to diverse functional groups to the cell surface (Fig. 40–2). As a result of mutant PIGA, progeny of the affected stem cells are deficient in all GPI-APs. Although more than 20 GPI-APs are expressed by hematopoietic cells, it is deficiency on red blood cells (RBCs) of the two GPI-anchored complement regulatory proteins, CD55 and CD59, that underlies the hemolytic anemia of PNH.17 RBCs lacking CD55 and CD59 undergo spontaneous intravascular hemolysis as a consequence of unregulated activation of the APC (see Fig. 40–1, bottom panel). Thus, the hallmark clinical manifestation of PNH (intravascular hemolysis and the resultant hemoglobinuria) occurs because the two proteins that regulate complement on erythrocytes happen to be GPI-anchored.
Figure 40–2.
The molecular and genetic basis of paroxysmal nocturnal hemoglobinuria (PNH). There are two types of anchoring mechanisms for plasma membrane proteins: transmembrane and glycosylphosphatidylinositol (GPI). Transmembrane proteins are anchored into the lipid bilayer of the cell by a short series (approximately 25 amino acids) of hydrophobic residues (blue rectangle). Transmembrane proteins typically have a short cytoplasmic tail that usually has signaling properties (red rectangle). The ectoplasmic portion of the protein is illustrated by the series of gray-blue squares. The GPI-anchored protein (AP) consists of the following components: phosphatidylinositol (inositol is represented by the blue hexagon labeled I and phosphate is represented by the red oval); glucosamine (GLcN, yellow circle); three mannose (Man, green circles); ethanolamine phosphate (EtN, blue square with attached phosphate represented by the red oval); the protein entity (blue circle). The lipid component (indicated by the series of diagonal lines within the lipid bilayer) is usually 1-alkyl,2-acylglycerol for mammalian GPI-APs. PNH cells are deficient in all GPI-APs because somatic mutation of the X-chromosome gene PIGA disrupts the first step in the biosynthetic pathway (transfer of the nucleotide sugar UDP-GlcNAc [uridine diphosphate–N-acetylglucosamine] to GlcNAc-PI [phosphatidylinositol]) indicated by the arrow.
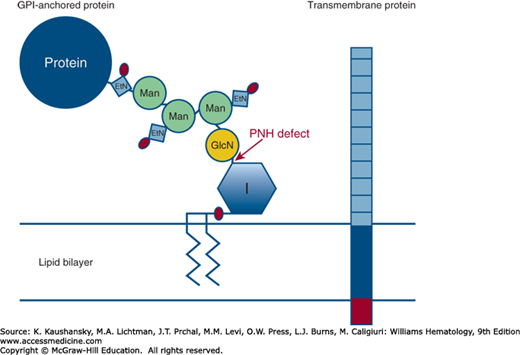
Hypothetically, the PNH phenotype would result from inactivation of any of the more than 25 genes involved in synthesis of the GPI-anchor (see Fig. 40–2), but, with one exception,18 somatic mutation of no gene involved in GPI-AP synthesis other than PIGA has been reported in patients with PNH. This phenomenon is accounted for by the fact that, of the genes involved in the GPI-anchor synthesis pathway, only PIGA is located on the X-chromosome. Therefore, somatic mutation of only one allele is required for expression of the phenotype as males have one X-chromosome and, as a consequence of X-inactivation during embryogenesis, females have only one functional X-chromosome in somatic tissues (Chap. 10). On the other hand, mutation of two alleles would be required for inactivation of any of the autosomal genes involved in the GPI-anchor synthesis pathway.18
Cells with PIGA mutations do not appear to have a proliferative advantage in vitro or in hybrid animal models made with PIGA knockouts.19 They have been found to be relatively resistant to apoptosis in some studies,20,21,22,23 but not in others.24,25 Thus, the basis of clonal selection and clonal expansion of PIGA mutant stem cells in patients with PNH remains largely enigmatic26 although a number of hypotheses have been proposed (reviewed in Ref. 17).
The blood of patients with PNH is a mosaic of normal and abnormal cells (Fig. 40–3). Although PNH is a clonal disease, the extent to which the PIGA-mutant clone expands varies widely among patients.17 As an example, in some cases, greater than 90 percent of the blood cells may be derived from the PIGA-mutant clone, whereas in others, less than 10 percent of the blood cells may be GPI-AP deficient. This unique feature (variability in extent of mosaicism) is clinically relevant because patients with relatively small PNH clones have minimal or no symptoms and require no PNH-specific treatment, whereas those with large clones are often debilitated by the consequences of chronic complement-mediated intravascular hemolysis and respond dramatically to complement inhibitory therapy.
Figure 40–3.
Phenotypic mosaicism is a characteristic feature of paroxysmal nocturnal hemoglobinuria (PNH). A. The blood of patients with PNH is a mosaic of phenotypically normal and abnormal cells. In some patients, erythrocytes that are partially deficient in glycosylphosphatidylinositol-anchored proteins (GPI-APs) (called PNH II) are present in the blood along with cells that are completely deficient (PNH III) and cells that are phenotypically normal (PNH I). In the case illustrated, erythrocytes from a patient with PNH (PNH, upper panels) and from a healthy volunteer (NL, lower panels) were stained with fluorescently labeled antibodies (anti-CD55, left panels; CD59, right panels) and analyzed by flow cytometry. B. PIGA genotype determines PNH phenotype. The PNH II phenotype is a consequence of PIGA mutation that partially inactivates enzyme function (red circles), whereas any PIGA mutation that causes complete loss of enzyme function generates the PNH III phenotype (green, yellow, and blue circles). PNH I cells, have wild-type PIGA and are the progeny of normal residual hematopoietic stem cells. In a single individual, multiple discrete PIGA mutations can be identified, accounting for the phenotypic mosaicism based on GPI-AP expression. DAF, decay-accelerating factor; MIRL, membrane inhibitor of reactive lysis.
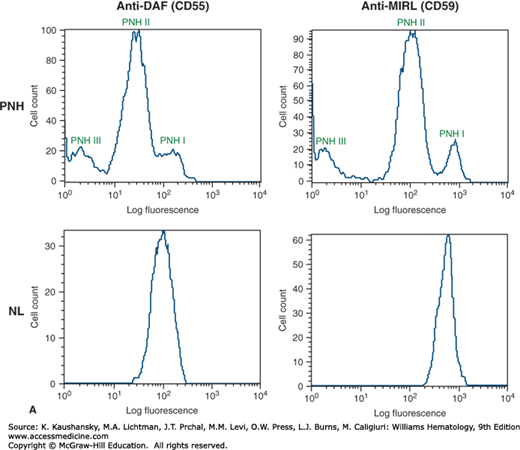
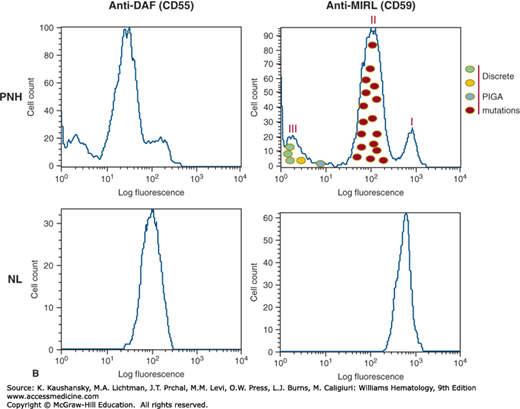
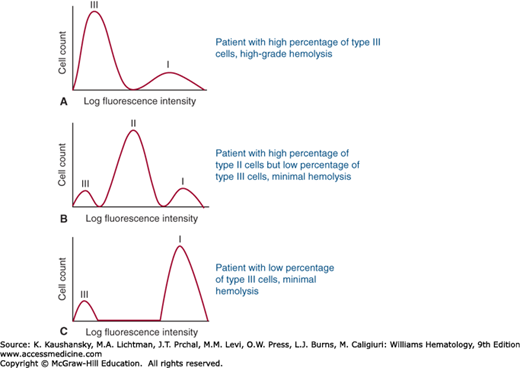
Another remarkable feature of PNH is phenotypic mosaicism (see Fig. 40–3A) based on PIGA genotype27 (see Fig. 40–3B) that determines the degree of GPI-AP deficiency.17 PNH III cells are completely deficient in GPI-APs, PNH II cells are partially (approximately 90 percent) deficient and PNH I cells express GPI-APs at normal density (putatively, these cells are progeny of residual normal stem cells; see Fig. 40–3A). Phenotype varies among patients (Fig. 40–4). Some patients have only type I and type III cells (the most common phenotype), some have type I, type II, and type III (the second most common phenotype), and some patients have only type I and type II cells (the least-common phenotype). Furthermore, the contribution of each phenotype to the composition of the blood varies. Phenotypic mosaicism is clinically important because PNH II cells are relatively resistant to spontaneous hemolysis, and patients with a high percentage of type II cells have a relatively benign clinical course with respect to hemolysis (Fig. 40–4).
Figure 40–4.
Clinical manifestations of paroxysmal nocturnal hemoglobinuria (PNH) are determined by clone size and erythrocyte phenotype. Mock flow cytometry histograms of erythrocytes from hypothetical patients with PNH stained with anti-CD59 are illustrated. Both the proportion and type of abnormal erythrocytes vary greatly among patients with PNH and these characteristics are important determinants of clinical manifestations. In general, patients with a high percentage of type III erythrocytes have clinically apparent hemolysis (A). If the erythrocytes are partially deficient in glycosylphosphatidylinositol-anchored protein (PNH II cells), hemolysis may be modest even if the percentage of the affected cells is high (B). A patient may have a diagnosis of PNH, but if the proportion of type III cells is low, only biochemical evidence of hemolysis may be observed (C). (Modified with permission from Parker C, Omine M, Richards S, et al: Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 106(12):3699–3709, 2005.)
The anemia of PNH is multifactorial as an element of marrow failure is present in all patients, although the degree of marrow dysfunction is variable.28 In some patients, PNH arises in the setting of aplastic anemia. In this case, marrow failure is the dominant cause of anemia. In other patients with PNH, evidence of marrow dysfunction may be subtle (e.g., an inappropriately low reticulocyte count) with the degree of anemia being determined primarily by the rate of hemolysis that is, in turn, determined by PNH clone size.
CLINICAL FEATURES
The primary clinical manifestations of PNH are hemolysis, thrombosis, and marrow failure.28 Constitutional symptoms (fatigue, lethargy, malaise, asthenia) dominate the history, with nocturnal hemoglobinuria being a presenting symptom in only approximately 25 percent of patients.29 Direct questioning frequently elicits a history of episodic dysphagia and odynophagia, abdominal pain, and male impotence. Venous thrombosis, often occurring at unusual sites (Budd-Chiari syndrome, mesenteric, dermal, or cerebral veins), may complicate PNH. Arterial thrombosis is less common.
LABORATORY FEATURES
PNH should be suspected in all patients with nonspherocytic, Coombs-negative intravascular hemolysis (Table 40–1).
History of episodic hemoglobinuria Evidence of nonspherocytic, Coombs-negative intravascular hemolysis (must have high serum lactate dehydrogenase) Patients with aplastic anemia (screen at diagnosis and once yearly even in the absence of intravascular hemolysis) Patients with refractory anemia or refractory cytopenias with multilineage dysplasia variants of myelodysplastic syndrome† Patients with venous thrombosis involving unusual sites (usually have evidence of intravascular hemolysis) • Budd-Chiari syndrome • Other intraabdominal sites • Cerebral veins • Dermal veins |
Although the clinical manifestations of PNH depend in large part on the size of the PIGA-mutant clone, the extent of the associate marrow failure also contributes significantly to disease manifestations. Thus, PNH is not a binary process and based on clinical features, marrow characteristics, and the size of the mutant clone as determined by the percentage of GPI-AP–deficient polymorphonuclear cells (PMNs), the International PNH Interest Group recognizes three disease subcategories (Table 40–2).28
| Category | Rate of Intravascular Hemolysis† | Marrow | Flow Cytometry | Benefit from Eculizumab |
|---|---|---|---|---|
| Classic | Florid (macroscopic hemoglobinuria is frequent or persistent) | Cellular marrow with erythroid hyperplasia and normal or near-normal morphology‡ | Large population (>50%) of GPI-AP–deficient PMNs¶ | Yes |
| PNH in the setting of another marrow failure syndrome§ | Mild to moderate (macroscopic hemoglobinuria is intermittent or absent) | Evidence of a concomitant marrow failure syndrome§ | Although variable, the percentage of GPI-AP deficient PMNs¶ is usually relatively small (<30%) | Dependent on the size of the PNH clone |
| Subclinical | No clinical or biochemical evidence of intravascular hemolysis | Evidence of a concomitant marrow failure syndrome§ | Small (<1%) population of GPI-AP–deficient PMNs detected by high-resolution flow cytometry | No |
Reticulocytosis reflects the response to hemolysis, although the reticulocyte count may be lower than expected for the degree of anemia because of underlying marrow failure (see Table 40–1
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


