Contemporary neuropathology plays a key role in the multidisciplinary management of brain tumor patients, in part due to increased supplementation of histopathological assessments by molecular diagnostic tests involving brain tumor tissue. Several molecular tests have become routine for clinical practice, and not only contribute to a refinement of tumor classification, but also aid in improved prediction of prognosis and in development of a tailored approach to therapy. This review provides an overview of classification and grading of brain tumors, particularly neuroepithelial tumors, and describes genetic/epigenetic changes that have gained clinical significance for molecular diagnostic testing.
- •
Contemporary neuropathology plays a key role in the multidisciplinary management of brain tumor patients.
- •
The histopathologic characterization and grading of brain tumors is increasingly supplemented by molecular diagnostic tests involving brain tumor tissue.
- •
Several molecular tests have become routine for clinical practice and not only contribute to a refinement of the standard classification scheme but also aid in improved prediction of prognosis and in the development of a tailored approach to clinical therapy.
Introduction
Primary brain tumors in adults are rare and constitute fewer than 2% of all malignant neoplasms. In children, however, they form the second most common type of tumor, after leukemias. Of all intracranial tumors, neuroepithelial tumors are the most common neoplasms, representing more than 60% of central nervous system (CNS)–derived lesions. Other lesions are meningothelial tumors (30%), tumors arising from cranial and spinal nerves (7%–8%), CNS lymphomas (4%), and germ cell neoplasms (approximately 1%). The classification system of the World Health Organization (WHO) lists more than 120 different tumor types and subtypes for the CNS ( Table 1 ). This review focuses on some of the main groups of neuroepithelial tumors, in particular glial neoplasms and embryonal tumors. Detailed discussion of neuronal, glioneuronal, pineal, and other tumor entities is beyond the scope of this article, especially because there is only limited molecular information available, which contributes to clinical management. This review is divided into 2 sections: section I describes the WHO classification, grading, epidemiology, and pathology of tumors, and section II focuses on molecular biology and prognostic markers, particularly of gliomas.
| WHO Grade | ||||
|---|---|---|---|---|
| I | II | III | IV | |
| Astrocytic Tumors | ||||
| Pilocytic astrocytoma | x | |||
| Pilomyxoid astrocytoma | x | |||
| Subependymal giant cell astrocytoma | x | |||
| Pleomorphic xanthoastrocytoma | x | |||
Diffuse astrocytoma
| x | |||
| Anaplastic astrocytoma | x | |||
Glioblastoma
| x | |||
| Oligodendroglial Tumors | ||||
| Oligodendroglioma | x | |||
| Anaplastic oligodendroglioma | x | |||
| Oligoastrocytic Tumors | ||||
| Oligoastrocytoma | x | |||
| Anaplastic oligoastrocytoma | x | |||
| Ependymal Tumors | ||||
| Subependymoma | x | |||
| Myxopapillary ependymoma | x | |||
Ependymoma
| x | |||
| Anaplastic ependymoma | x | |||
| Choroid Plexus Tumors | ||||
| Choroid plexus papilloma | x | |||
| Atypical choroid plexus papilloma | x | |||
| Choroid plexus carcinoma | x | |||
| Embryonal Tumors | ||||
Medulloblastoma
| x | |||
CNS PNET
| x | |||
| Atypical teratoid/rhabdoid tumor | x | |||
| Neuronal and mixed Neuronal-glial Tumors | ||||
| Gangliocytoma | x | |||
| Ganglioglioma | x | |||
| Anaplastic ganglioglioma | x | |||
| Desmoplastic infantile astrocytoma and ganglioglioma | x | |||
| Dysembryoplastic neuroepithelial tumor | x | |||
| Central neurocytoma | x | |||
| Extraventricular neurocytoma | x | |||
| Cerebellar liponeurocytoma | x | |||
| Paraganglioma of the spinal canal | x | |||
| Papillary glioneuronal tumor | x | |||
| Rosette-forming tumor of the fourth ventricle | x | |||
| Other Neuroepithelial Tumors | ||||
| Chordoid glioma of the third ventricle | x | |||
| Angiocentric glioma | x | |||
Section I: pathologic classification, epidemiology, and morphology of neuroepithelial tumors
WHO Classification and Grading of Tumors
Brain tumors are classified according to international standards, which are established by the WHO. The WHO classification of CNS tumors is based on consensus criteria developed by an international working group of pathologists and geneticists and forms a comprehensive worldwide standard for tumor definition. The most recent update was published in 2007 (4th edition) and led to an integration of several new histologic subtypes and a revision and expansion of molecular genetic data.
Brain tumors are primarily classified according to their histopathologic appearance, which is based on the characterization of constituent cell types and growth patterns. Although the cellular origin of many tumors is still under investigation, the histologic classification relies on morphologic similarities of tumor cells with their presumed non-neoplastic counterpart, in addition to the presence of certain architectural patterns. The WHO subdivides the large group of neuroepithelial neoplasms into a variety of histologic tumor types, such as glial (astrocytomas, oligodendrogliomas, and ependymomas), neuronal, mixed glial-neuronal, embryonal, pineal, choroid plexus derived and others (see Table 1 ). Many molecular profiles have been incorporated and contribute to the subclassification of tumor entities.
The WHO classification also has devised a grading scheme, which can be considered a malignancy scale that is used to predict the biologic behavior of neoplasms, therapeutic response, and clinical outcome. It is mainly based on histologic criteria, such as cell density, infiltrative nature, nuclear pleomorphism, mitoses, vascular proliferation, and necrosis. The grading of tumors follows a 4-tiered system and is designed to add a numeric value to the histologic grade (see Table 1 ), which ranges from I (benign) to IV (malignant). Grade I is assigned to tumors that are circumscribed, show a low proliferative potential, rarely progress to malignancy, and are likely cured with surgical resection alone. Grade II tumors have a low proliferative potential but tend to be infiltrative in nature and often recur. Many of these lesions progress to a higher grade of malignancy over time. Grade III lesions show histologic evidence of malignancy, such as nuclear atypia and high proliferative activity. These lesions cannot be cured with surgery alone and require adjuvant radiation and/or chemotherapy. Grade IV is reserved for frankly malignant tumors with an aggressive preoperative and postoperative course. Histologically, these lesions demonstrate pronounced nuclear atypia, high mitotic activity, vascular proliferation, and a tendency to necrose.
The WHO histologic grade, however, is only one of many factors that aid in predicting response to therapy and outcome. Additional, clinically important attributes are a patient’s age and neurologic performance status, tumor location, and extent of surgical resection as well as radiologic aspects, such as tumor contrast enhancement. Molecular genetic alterations and proliferation indices play another major role. Combinations of these factors are used to provide an estimate of prognosis and outcome for each tumor entity. On average, patients with WHO grade II tumors tend to survive for greater than 5 years, whereas the survival time of patient’s with grade III lesions is reduced to 2 to 3 years. WHO grade IV tumors are in general rapidly fatal if untreated (survival <1 year), and survival rates are greatly influenced by effective adjuvant treatment regimens.
Astrocytic Tumors
Astrocytomas are a morphologically heterogeneous group of neoplasms and defined as tumors with predominantly astrocytic differentiation. They are divided into approximately 2 main groups: (1) diffuse astrocytic lesions, which are more common and biologically related, and (2) circumscribed astrocytic lesions, which are less frequent and biologically unrelated. Diffuse astrocytic tumors are further subdivided according to their degree of malignancy and range from WHO grade II (diffuse astrocytomas and variants), to WHO grade III (anaplastic astrocytomas), to WHO grade IV (glioblastoma and variants). The more circumscribed astrocytic tumors include pilocytic astrocytoma (WHO grade I), pleomorphic xanthoastrocytoma (WHO grade II), subependymal giant cell astrocytoma (WHO grade I), and pilomyxoid astrocytoma, a recently added variant, which is considered WHO grade II. These circumscribed tumors have an indolent course and rarely progress to malignancy.
Diffuse astrocytomas
Diffusely infiltrating astrocytic tumors are the most common primary neoplasms in adults and constitute more than 60% of all brain tumors. The incidence of astrocytomas differs somewhat regionally, but recent estimates suggest an incidence rate of 0.4 per 100,000 people for grade II astrocytomas and an incidence rate of 3.2 per 100,000 people for glioblastoma. The histologic grade shows a direct correlation with the age at presentation, whereby WHO grade II tumors present in younger adults in their fourth or fifth decades, whereas glioblastomas (WHO grade IV) peak in the elderly (mean age at diagnosis 61 years). Men seem more affected than women with a male-to-female ratio of 1.5:1.0 for all astrocytic tumors (except pilocytic astrocytoma).
WHO grade II diffuse astrocytomas are morphologically heterogeneous and characterized by a high degree of cellular differentiation, slow growth, low mitotic activity, and diffuse infiltration and spread into adjacent brain structures ( Fig. 1 ). Tumor cells express glial fibrillary acidic protein (GFAP), a protein typically found in astrocytomas. WHO grade II diffuse astrocytomas can be found at any site in the CNS but preferentially in the cerebral hemispheres, particularly in the subcortical and deep white matter of frontotemporal lobes. Although these lesions are rare in children, the main site in pediatric patients is the brainstem (so-called brainstem glioma). Three major variants of WHO grade II diffuse astrocytomas can be distinguished morphologically, and these comprise the fibrillary, gemistocytic, and protoplasmic variants. These subtypes, however, often coexist and a clear subclassification is not possible.
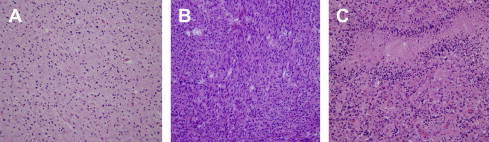
Anaplastic astrocytomas (WHO grade III) are defined as diffuse astrocytomas with focal or dispersed anaplasia. They may arise from low-grade astrocytomas but are also frequently diagnosed at first biopsy, without indication of a less malignant precursor lesion. These tumors are grossly more discernible because they are more cellular and form a more readily identifiable tumor mass. The infiltrative nature tends to create an overall increase of tissue volume without inducing a destructive effect. In comparison to low-grade tumors, these neoplasms are microscopically remarkable for increased cellularity and enlarged, irregular hyperchromatic nuclei (see Fig. 1 ). Capillaries are lined by a single layer of endothelium, and frank vascular proliferation and necrosis are not present. Immunoreactivity for GFAP is less consistent than for grade II lesions. In contrast to low-grade astrocytomas, these lesions display increased mitotic activity with a proliferative fraction (Ki-67/MIB-1 labeling) of 5% to 10%.
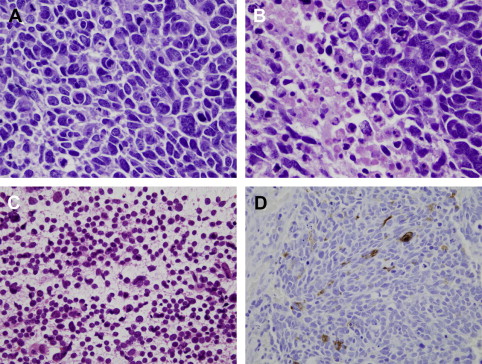
Glioblastomas (WHO grade IV) are the most malignant tumors within the spectrum of diffuse astrocytoma and account for approximately 12% to 15% of all intracranial neoplasms and up to 60% of all astrocytic tumors. They affect mainly adults with a peak incidence between 40 and 70 years. Fewer than 10% of glioblastomas arise from a lesion of lower malignancy grade (secondary glioblastoma) and manifest in younger patients (mean age of 45 years). Most are found de novo (primary glioblastoma) after a short clinical history and are seen in older individuals (mean age 62 years). The majority of tumors are located within the cerebral hemispheres; they often infiltrate deep nuclei and spread along white matter tracts to the contralateral hemisphere. Brainstem involvement is rare and mainly present in children. Other sites, such as spinal cord or cerebellum, are infrequently involved. Microscopically, glioblastomas are heterogeneous. They show a high degree of cellularity, pleomorphism, and mitotic activity in addition to microvascular proliferation and necrosis (see Fig. 1 ). The latter two features are the cardinal diagnostic features of glioblastomas and help distinguish them from grade III astrocytomas. Three distinct glioblastoma variants have been described, which are gliosarcoma, giant cell glioblastoma, and small cell glioblastoma.
Gliosarcomas are defined as glioblastoma intermixed with a sarcomatous component and represent approximately 2% of all glioblastomas. The clinical features are similar to those of classic glioblastomas. Critical diagnostic parameters are a biphasic growth pattern with areas of glial and mesenchymal differentiation. Molecular changes are variable but similar to those occurring in glioblastoma; however, tumor histogenesis is controversial.
Giant cell glioblastomas are remarkable for the presence and predominance of markedly bizarre, multinucleated giant cells, within an abundant stromal reticulin network. In spite of their unusual appearance, the consistent expression of GFAP in conjunction with genetic profiling confirmed its astrocytic nature.
Small cell glioblastoma is characterized by a predominance of small, anaplastic cells that resemble those in a primitive neuroectodermal tumor (PNET). The distinction is made via immunohistochemistry, because these tumors lack expression of synaptophysin that is typical of PNET. The small cell phenotype seems to correlate with the presence of EGFR amplification. Although typical glioblastoma features are not present, overall survival times are less than 1 year.
Circumscribed astrocytic lesions
Pilocytic astrocytomas are slowly growing, well circumscribed glial tumors with preferential growth in midline CNS structures. More than 75% of these tumors are seen in children and adolescents within the ages of 8 to 13 years. These tumors represent approximately 6% of all intracranial tumors and occur at an incidence of less than 1 case per 100,000 individuals per year. In infancy it is one of the most frequent brain tumors. The cerebellum is the most commonly affected site, with preferential involvement of the cerebellar hemispheres. The optic nerve and chiasm are second in frequency, followed by the cerebral cortical hemispheres, brainstem, infundibulum, hypothalamus, and spinal cord. In the brainstem, these lesions tend to occur mainly in the dorsal regions and pontomedullary junction, contrasting diffusely infiltrative astrocytomas, which grow typically in the ventral pons. The histopathology can show a variety of patterns and pose great diagnostic challenges. PAs typically demonstrate a biphasic pattern in which fibrillated, compact areas are intermingled with loosely structured, microcystic, and mucinous regions within the tumor tissue. The predominant cell type is an elongated, unipolar or bipolar cell with thin hair-like (Greek pilos [hair]) processes, which is immunoreactive for GFAP. The histogenetic origin of these tumors is still enigmatic. Characteristic of PAs is the presence of Rosenthal fibers and eosinophilic granular bodies. These are bright eosinophilic, either carrot-shaped or round structures, and considered amorphous degradation products associated with intermediate filaments. Mitoses are in general absent or infrequent, and the proliferative index (Ki-67/Mib-1) is less than 1%, although it can be focally slightly elevated. PAs have a tendency to invade leptomeninges or subarachnoid space. In contrast to diffusely infiltrating astrocytomas, PAs do not have an intrinsic tendency to progress to a higher grade and are considered a benign glial tumor. The survival rates range from close to 100% at 5 years to approximately 95% at 10 years with recurrence-free intervals of up to 20 years. The WHO assigned grade I to PAs, whereby the grading scheme developed for diffusely infiltrating astrocytomas does not apply. Rarely, these lesions show more aggressive biologic behavior characterized by significant proliferative activity; they are then considered WHO III tumors, equivalent to anaplastic astrocytomas.
Pilomyxoid astrocytoma is considered a variant of pilocytic astrocytoma and typically found in the hypothalamic/chiasmatic region. Histologically, pilomyxoid astrocytoma is composed of monomorphic, bipolar cells with angiocentric arrangements and a striking mucinous background. In contrast to pilocytic astrocytomas, these tumors do not display a biphasic cytoarchitecture; furthermore, they lack Rosenthal fibers or eosinophilic granular bodies and show a high mitotic rate. Infants and children (median age 10 months) are the main affected groups, and prognosis seems less favorable than for PAs. Local recurrences and spread through cerebrospinal fluid do occur more readily than with pilocytic astrocytomas; thus, the WHO assigned grade II to these tumors.
Pleomorphic xanthoastrocytomas are rare tumors, which account for fewer than 1% of glial neoplasms and typically manifest in children and young adults, in whom they are associated with a longstanding history of seizures. The majority of these tumors are found in a superficial location, particularly in the temporal lobe, and there is a tendency to involve the overlying subarachnoid space. Characteristic histologic features are multinucleated and lipidized giant cells with bizarre nuclei, which tend to express GFAP, an astrocytic marker. A reticulin rich stromal background is a hallmark feature. These tumors show low proliferative activity. Macroscopically, they are well demarcated and only small portions seem to invade adjacent brain and perivascular spaces. These tumors have been assigned WHO grade II. Many pleomorphic xanthoastrocytomas show a benign clinical course, usually with long-term survival after tumor resection. Only few tumors undergo malignant transformation.
Subependymal giant cell astrocytomas are benign neuroepithelial tumors, which tend to develop in the context of tuberous sclerosis. These tumors typically manifest before age 30 years and are located within the walls of the lateral ventricles. Histologically, they are composed of large ganglioid cells with both astrocytic and neuronal differentiation. Their histogenesis, however, is largely unknown. It is hypothesized that they are related to and/or derived from hamartomatous lesions typically seen in tuberous sclerosis. Subependymal giant cell astrocytomas are designated WHO grade I. Symptomatic lesions are treated with resection and many studies have shown that these tumors have an almost uniformly favorable prognosis.
Oligodendroglial Tumors
Oligodendrogliomas form a group of diffusely infiltrative glial tumors with unique pathologic, clinical, and molecular characteristics. They are composed of neoplastic cells with features reminiscent of oligodendrocytes and often harbor deletions of the short arm of chromosome 1 (1p) and the long arm of chromosome 19 (19q).
Oligodendrogliomas account for approximately 5% to 6% of all glial neoplasms and for 2% to 3% of all primary brain tumors. The annual estimated incidence rate lies in a range of 0.27 to 0.35 per 100,000 individuals. Although oligodendrogliomas can develop at any age, the majority of tumors arise in the fourth to fifth decade, and fewer than 2% of oligodendrogliomas are found in children younger than 14 years, with a slight male predominance. Oligodendrogliomas can arise anywhere within the CNS, but the majority of tumors are found within the frontal and temporal lobes of the cerebral hemispheres. They are present to a lesser extent in other cortical regions and they are rare within the deep nuclei or spinal cord. Histologically, these tumors are composed of a monomorphous population of cells with round, regular nuclei, cytoplasmic clearing (on routinely formalin-fixed paraffin embedded sections), and growth in close proximity to fine branching vasculature ( Fig. 2 ).
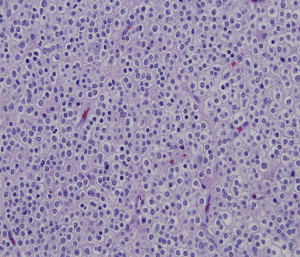
The WHO classification divides the spectrum of oligodendrogliomas into well-differentiated tumors, which are slow-growing neoplasms and histologically correspond to WHO grade II and anaplastic tumors, which are WHO grade III. Anaplastic oligodendrogliomas are characterized by a high degree of anaplasia, an increase in cellularity, nuclear pleomorphism, in addition to brisk mitotic activity, endothelial proliferation, and necrosis. In contrast to other gliomas, like astrocytomas and ependymomas, oligodendrogliomas show a more slowly progressive clinical course and demonstrate chemosensitivity. Their molecular signature is distinct and features codeletions of 1p and 19q. Classic oligodendroglial histology in conjunction with 1p/19q codeletions has been strongly linked to more favorable clinical behavior and is considered a defining constellation (discussed later).
Mixed Oligoastrocytomas
Oligoastrocytomas are defined as diffusely infiltrative glial neoplasms consisting of a mixture of 2 distinct neoplastic cell types, which morphologically resemble the tumor cells of diffuse astrocytomas as well as oligodendrogliomas. These 2 components coexist either side-by-side or in a diffusely intermingled fashion. Definitive criteria for identification and classification of these lesions, however, remain somewhat controversial. Oligoastrocytomas are graded as WHO grade II lesions and the acquisition of anaplastic features increase the grade to WHO grade III.
Ependymal Tumors
Ependymomas are defined as slowly growing glial neoplasms, which can arise anywhere along the walls of the cerebral ventricles or within the spinal canal. The group of ependymal tumors comprises classic ependymoma (plus variants), anaplastic ependymoma (malignant variant), and the benign variants, subependymoma and myxopapillary ependymoma.
Ependymomas account for approximately 5% to 6% of all gliomas and for 2.5% of all primary intracranial neoplasms in adults. In children younger than 14 years, these tumors play a significant role and form approximately 7% to 8% of all primary intracranial neoplasms, with an adjusted annual incidence rate of 5 to 6 per 1 million individuals. Overall, ependymomas are the third most common pediatric tumor after astrocytomas and medulloblastomas. Ependymomas can develop at any age; however, there are 2 distinct incidence peaks, the first one in children before the age of 14 years and the second one in adults between 35 and 45 years. These tumors can arise anywhere along the ventricular system within brain and spinal canal, but approximately 60% of lesions are located in the fourth ventricle, particularly in pediatric patients. In the spinal cord it is the most common type of glial neoplasm affecting adults. Male are in general slightly more affected than female.
Morphologically, classic ependymomas are composed of a monotonous population of cells, which tend to form characteristic rosette-like structures, so-called perivascular pseudorosettes, and ependymal rosettes that have been recognized as diagnostic hallmark features ( Fig. 3 ). Recent studies suggest that they might arise from radial glial cells.
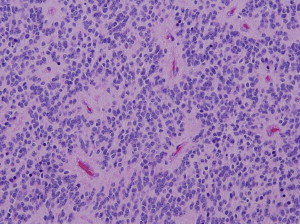
The WHO classification separates ependymal tumors into 3 grades, wherein subependymoma and myxopapillary ependymoma correspond to WHO grade I, classic ependymoma and related variants (cellular, papillary, clear cell, and tanycytic ependymoma) correspond to WHO grade II, and anaplastic ependymomas are WHO grade III.
Anaplastic ependymomas are the malignant variant of classic ependymomas, characterized by high cell density, high mitotic activity, microvascular proliferation, and necrosis. Anaplastic ependymomas are associated with rapid disease progression and unfavorable outcome.
Myxopapillary ependymoma is a distinct type of low-grade ependymoma, which is almost exclusively located in the conus medullaris, cauda equina, and filum terminale. These lesions are circumscribed and composed of small cuboidal cells surrounding well-vascularized, acellular cores of connective tissue, which frequently undergo hyaline and mucoid degeneration, leading to a distinctive histologic appearance. Myxopapillary ependymomas are slow-growing tumors with an indolent clinical course and favorable outcome after surgical resection, thus the WHO assigned grade I.
Subependymomas are benign neoplasms, typically located within the ventricular wall and composed of clusters of uniform small tumor cells. These are embedded in a densely fibrillar glial matrix with microcystic degeneration. Subependymomas correspond to WHO grade I.
Embryonal Tumors
Embryonal tumors of the CNS are primitive neuroectodermal tumors (PNETs), which are composed of undifferentiated small cells with high nuclear-to-cytoplasmic ratio and a lack of distinctive architectural features ( Fig. 4 ). This tumor group includes medulloblastoma (and variants), CNS PNETs, atypical teratoid/rhabdoid tumor, CNS neuroblastoma, CNS ganglioneuroblastoma, medulloepithelioma, and ependymoblastoma. The basis for grouping these lesions relates to their biologic tendency to differentiate into glial and neuronal lineages.