Drug name
Generic name
Target
Status
Reversible
ZD1839
Gefitinib
EGFR
Approved
OSI776
Erlotinib
EGFR
Approved
BPI-2009H
Icotinib
EGFR
Approved
TAK-165
Mubritinib
EGFR/ERBB2
Phase I
XL647
NA
EGFR/ERBB2/FLT-4
Phase II
ZD6474
Vandetanib
EGFR/ERBB2/RET
Phase III
GW572016
Lapatinib
EGFR/ERBB2
Preclinical
Irreversible
EKB-569
Pelitinib
EGFR
Phase I
CI-1033
Canertinib
EGFR/ERBB2/ERBB4
Phase II
HKI-272
Neratinib
EGFR/ERBB2
Phase II
BIBW2992
Afatinib
EGFR/ERBB2/ERBB4
Phase III
PF-00299804
Daconitinib
EGFR/ERBB2/ERBB4
Phase III
Third generation
CO-1686
NA
EGFR T790M
Phase I/II
WZ4002
NA
EGFR T790M
Preclinical
Second Generations Inhibitors
The development of drugs that bind irreversibly to ERBB family members and/or inhibit multiple targets simultaneously, are being investigated to treat NSCLCs that are resistant to first-generation EGFR TKIs [11]. Unlike reversible TKIs, irreversible TKIs contain a reactive Michael-acceptor group that binds covalently with Cys797 present at the ATP-binding cleft of mutant EGFR. This approach provides greater presence at the ATP site and overcoming the competition with ATP [6, 38]. The ability of an irreversible TKI to overcome resistance was demonstrated in vitro in mutant EGFR cell lines [39]. Several investigational irreversible multitargeted HER family TKIs (Table 1) are being evaluated in patients with NSCLC. These include neratinib or HKI-272 (Wyeth, which was acquired by Pfizer in 2009, New London, CT), PF00299804 (Pfizer), and afatinib or BIBW 2992 (Boehringer Ingelheim, Ingelheim, Germany).
Neratinib (HKI-272)
Neratinib , an irreversible ERBB family inhibitor that targets EGFR/ERBB1, ERBB2, and ERBB4 [40, 41] (Table 1), was evaluated in a phase I trial of patients with advanced solid tumors [42]. Of 14 evaluable patients with NSCLC, stable disease (SD) for 24 weeks was observed in six (43 %) patients. Despite preclinical data suggesting a role for neratinib in overcoming resistance mediated by T790M [39], no patients with a known T790M mutation responded in another study. Based on overall results, neratinib is no longer in development for NSCLC (http://www.clinicaltrials.gov), although it is being investigated in ERBB-2 positive breast cancer [43].
PF00299804
PF00299804, an irreversible ERBB family inhibitor that targets EGFR/ERBB1, ERBB3, and ERBB4 [44] (Table 1), has demonstrated preclinical activity in gefitinib-resistant NSCLC models both in vitro and in vivo [45]. In a phase I/II trial of PF00299804 in patients with NSCLC who progressed following one or two prior chemotherapy regimens and erlotinib [46], 36 patients with adenocarcinoma and five patients with non-adenocarcinoma histology were evaluated for efficacy. Among patients with adenocarcinoma, 67 % had a clinical benefit (response), and among those with non-adenocarcinoma histology, the clinical benefit rate was 40 %.
Afatinib (BIBW 2992)
Afatinib is an oral irreversible ERBB family inhibitor that targets EGFR/ ERBB1, ERBB2 [47], and ERBB4 with preclinical data supporting a role in overcoming resistance to reversible EGFR TKIs [47]. Afatinib has been studied in multiple phase I clinical trials [47–52]. Three patients with NSCLC experienced PRs lasting 24, 18, and 34 months; their tumors were found to have mutations in EGFR, although none had received prior EGFR TKI treatment. Two additional patients (one with NSCLC and one with esophageal cancer) had unconfirmed partial responses (PRs). One of the NSCLC patients with an activating exon 19 mutation who had a PR was initially treated with afatinib (10 mg/day) but subsequently progressed and developed brain metastases. By investigator assessment, the objective relative risk (RR), disease control rate (DCR), median progression free survival (PFS) interval, and median OS time were 60 %, 86 %, 14 months, and 24 months, respectively, for all patients [49]. The objective RR, DCR, and median PFS were 59 %, 83 %, and 16.1 months, respectively, for patients with L858R mutations and 69 %, 93 %, and 13.7 months, respectively, for patients with exon 19 deletions.
Third Generation (T790M EGFR Specific) Tyrosine Kinase Inhibitors
Despite the initial promise, the 4-anilinoquinazoline core structure that is common to the clinically available irreversible inhibitors, the second generation TKIs, do not show specificity towards T790M EGFR compared with wild-type EGFR. New structurally distinct irreversible ERBB family inhibitors, such as the pyrimidine-based inhibitors described were recently introduced by Zhou et al. [53]. This study screened a library of compounds to identify agents that inhibited growth of gefitinib-resistant and gefitinib-sensitive cell lines without producing toxicity in mutant KRAS cells at high concentrations. One such compound, WZ4002, is an irreversible inhibitor with chemical properties that favor 100-fold greater binding to the T790M mutant. WZ4002 also demonstrated a 100-fold weaker binding to wild-type EGFR than with neratinib and other quinazoline-based second generation EGFR inhibitors. Additionally, WZ4002 inhibited L858R/T790M EGFR kinase activity more potently than wild-type EGFR protein activity, whereas the opposite was true for neratinib and gefitinib. Such findings indicate that the concept of irreversible ERBB family inhibition is a very promising and may yet provide a solution to the problem of acquired resistance.
cMET Inhibitors
Multiple agents that inhibit the cMET signaling at various points have been studied. HGF-competitive analogs, such as NK4, have shown inhibitory activity in various cancer cell lines, [54–59]. Other compounds such as decoy cMET and the isolated Sema domain of cMET have the ability to simultaneously bind to both the ligand HGF and the receptor cMET [60, 61]. These agents have shown inhibition of cMET signaling in preclinical studies. Studies with specific antibodies against HGF/cMET have also shown encouraging results. Monoclonal antibodies against cMET, such as OA-5D5 and DN30, have been shown to cause tumor-cell growth inhibition [62–64]. In addition, monoclonal antibodies against HGF have also been developed (L2G7 and AMG102) and validated in several preclinical studies [65, 66]. Another way to inhibit the cMET pathway is through competitors for the ATP binding site in the TK domain of cMET. This type of inhibition is carried by the small molecule inhibitors such as PHA665752, ARQ 197, SGX523, JNJ-38877605, EXEL-2880, XL-184, MGCD265, MK2461, crizotinib (PF-02341066), K252a and MP470 (Table 2). Engelman et al. exposed the gefitinib resistant cells to the cMET inhibitor PHA665752, and restored tumor sensitivity to gefitinib by reducing EGFR phosphorylation and inducing apoptosis [15, 67]. In the model of immnodeficient mice with enhanced tumor growth due to increased production of HGF, the MET-specific small-molecule kinase inhibitor SGX523 partially inhibits the HGF-dependent growth of lung, breast and pancreatic tumors [68]. So far, the most relevant compound developed in this arena is crizotinib (PF02341066), which has been recently approved by the US FDA for the treatment of EML4/ALK mutant NSCLC tumors, targets cMET and ALK receptor TK. In a panel of different tumor cell lines, crizotinib inhibited phosphorylation of wild-type cMET. In lung carcinoma cells, crizotinib inhibited HGF-stimulated cell migration and invasion [69].
Table 2
cMET inhibitors under development
Agent | Mechanism of action |
|---|---|
PHA665752 | Specific cMET inhibitor, ATP competitor |
ARQ197 | Selective cMET inhibitor, non-ATP competitor |
SGX523 | Selective cMET inhibitor, ATP competitor |
JNJ-38877605 | Selective cMET inhibitor, ATP competitor |
EXEL-2880 | TKI, targets HGF and VEGFR family members |
XL-184 | Pan TKI |
MGLD-265 | cMET, VEGFR 1/2/3, Tie 2, Ron |
MK-2461 | Multi TKIs |
K252a | Multi TKIs |
MP470 | TKI targets cMET, PDGFR, c-Kit |
Strategic Combination Therapies
In selected cases combination therapies have been utilized, specifically to inhibit the compensatory pathway which is activated and mediates TKI resistance.
Combining EGFR and c-MET Inhibitors
For tumors harboring cMET amplification as a determinant to cause TKI resistance, targeting cMET receptor in combination to EGFR inhibitors are likely to predict a better response compared with individual targeting of EGFR. Antibodies targeting the cMET ligand, antibodies targeting MET itself and small molecules inhibitors against cMET are the therapeutic options for these combination therapies. The simultaneous inhibition of EGFR and cMET was shown to suppress the proliferation of cells and anti-tumor efficacy in mice in HCC827 cells which develop gefitinib resistance [15, 17]. Another study was carried out in NCI-H820 cells which naturally harbor EGFR T790M mutation as well as cMET amplification. In this study, small molecule cMET inhibition or knockdown of cMET along with EGFR inhibition suppressed the compensatory ERBB3 signaling and compromised cell viability [14]. From these studies, it was not clear whether T790M EGFR and cMET amplification co-occur in the same cell and whether the cell type is dependent on both of these factors. To address these issues, Xu et al. carried out a study, where they developed mouse model of adenocarcinoma harboring both T790M EGFR and cMET. In this study treatment with individual inhibitors of EGFR and cMET was un-affective and the combinatorial targeting of both these receptors caused significant tumor regression. Importantly, this study strengthened the notion that in tumors harboring both T790M and cMET amplifications, both these lesions are drivers for growth [70].
EGFR and ERBB2 Inhibitors
The rationale of combining EGFR and ERBB2 inhibitors is via various molecular interactions across their downstream signaling pathways. It is known that the ligand independent activation of EGFR can be mediated by ERBB2 amplification. Over-expression and amplification of ERBB2 decreased the degradation of EGFR and increases its recycling to the cell membrane [71]. For simultaneous targeting of EGFR and ERBB2, two classes of inhibitors have been developed: agents which bind reversibly and those that bind irreversibly (covalently) to the ATP binding site in the tyrosine kinase domain in EGFR and ERBB2. Due to the mechanisms governing the resistance to reversible TKI, irreversible inhibitors targeting both EGFR and ERBB2 are likely to be better therapeutic choices. Irreversible TKI such as BIBW 2992 is shown to inhibit the autophosphorylation of EGFR and ERBB2. This agent was more than 100-fold potent that gefitinib against cells harboring the T790M+L858R mutation [47]. Another irreversible inhibitor HKI-272 caused dramatic tumor regression in mice model of TKI resistance [72]. In a Phase I clinical trial of BIBW2992, out of 26 patients with adenocarcinoma, 2 showed partial response. Another Phase II single arm clinical trial using BIBW2992, recently reported partial response in 43 patients out of 67 in mutation positive patients. The disease control rate was 96 % and a median progression free survival was 10.2 months. A clinical trial utilizing HKI-272 showed stable disease in 42 % of 16 NSCLC patients previously treated with gefitinib.
EGFR and EMT Inhibitors
To overcome the EMT associated with TKI resistance several mediators have been targeted [73]. Due to the role of ERK1/2 in EMT, ERK1/2 blockade by U0126 led to a suppression of TGF-Beta mediated EMT in NSCLC, restored epithelial phenotype of these cells and sensitized the TKI resistant cells in combination with gefitinib [74]. E-cadherin re-expression is also utilized to target EMT in overcoming TKI resistance. ZEB1 is known to down regulate E-cadherin and promote EMT. ZEB1 was shown to be inhibited by utilizing Histone deacetylase inhibitors. In this study, the combination of HDAC inhibitors with erlotinib led to the reversal of TKI resistance in HCC4006ER cells [19]. Since the up regulation of AXL kinase was shown to be a critical determinant of TKI resistance in NSCLC, targeting of AXL kinase sensitized HCC827ER cells. In this study, knockdown of AXL kinase in HCC827 parental cells did not affect survival, however, the knockdown decreased the survival of HCC827ER cells, suggesting the specific role of AXL kinase in mediating erlotinib resistance in these cells. Small molecule inhibitors of AXL kinase, MP-470 and XL-880 [28] in combination with erlotinib also decreased the viability of HCC827ER cells. The process of EMT is also governed by inflammatory signals [75]. The inflammatory enzyme COX-2 is frequently over-expressed in a variety of malignancies [76]. COX2 plays an important role in conferring malignant and metastatic phenotypes and its over-expression is involved in therapy resistance [77]. For instance, COX-2 over-expression in NSCLC is associated with apoptosis resistance, [78] angiogenesis, [79, 80] and metastasis [81, 82]. Most of these effects are mediated by prostaglandin E2 (PGE2). PGE2 and other inflammatory cytokines in the tumor microenvironment may contribute to TKI resistance by downregulating the levels of E-cadherin. These findings suggest a strong rationale of combining COX inhibitors with TKI to overcome TKI resistance [83]. However the clinical trials combining COX2 inhibitors with EGFR inhibitors did not show additional benefits compared with the individual treatment of EGFR inhibitor. It seems possible that in these trials sufficient dose of COX2 inhibitor was not used to inhibit maximum COX2 activity [84, 85].
EGFR and mTOR Pathway Inhibitors
The rationale for combining EGFR inhibitors with mTOR inhibitors was based on studies suggesting an important survival function of mTOR-AKT axis as downstream effector of EGFR signaling [86]. The ability of the irreversible EGFR inhibitor HKI-272 and rapamycin combination to promote more effective suppression of EGFR signaling to S6 and AKT kinases was demonstrated both in cultured NSCLC cell lines harboring double mutant EGFR alleles as well as in lung tumors in TL mice. The investigators suggest that HKI-272 may not sufficiently overcome the biochemical drug resistance conferred by T790M and that further suppression of an essential AKT-mTOR signal downstream of EGFR is required to achieve a therapeutic response [87].
Promoting Oncogene Degradation
An interesting aspect by which TKI resistance has been targeted is via EGFR degradation. It is now well accepted that EGFR degradation is superior in causing cancer cell cyto-toxicity compared to simply inhibiting its tyrosine kinase activity. Weihua et al. reported that knockdown of EGFR caused autophagy and inhibition of EGFR only caused a transient cell cycle arrest [88]. Other findings in head and neck tumor models suggest that promoting EGFR degradation may lead to cell death [89, 90]. Chemotherapeutic agents such as gemcitabine and cisplatin caused EGFR degradation which correlated with cell death. The importance of targeting EGFR for degradation to overcome TKI resistance was shown by knocking down EGFR in several EGFR mutant cell lines. In this study, knockdown of EGFR caused a decrease in survival of EGFR dependent cells including NCI-H1975 cells harboring EGFR-T790M mutation [91]. This approach was utilized recently to develop a peptide based therapy which caused EGFR degradation in TKI resistant cells harboring T790M mutant EGFR. The peptide, named as Disruptin was shown to decrease survival of TKI resistant NCI-H1975 cells by disrupting EGFR-Heat shock protein 90 interaction, inhibiting EGFR homodimerization and promoting EGFR degradation [92].
Conclusions and Future Perspectives
The main signaling pathways contributing towards TKI resistance and the therapeutic approaches to rationally target them are summarized in Fig. 1. In order to better target the EGFR TKI resistance, it will be critical to understand the driver oncogene mediating resistance in a specific patient. The prescreening of patients for the driver mutations and amplifications need to be carried out and the personalized therapies need to be tailored depending upon the driver oncogene. For example, in patients with AXL kinase amplification or COX2 over-expression, AXL or COX2 inhibitors need to be combined with EGFR TKI. Several preclinical studies suggest the re-emission of resistance due to another compensatory mechanism in response to individual therapies. As for instance, in case of irreversible EGFR inhibitors, resistance develops due to downstream signaling mediators. A more effective approach would be to combine the first generation EGFR inhibitor with the second or preferably third generation TKIs. Another promising approach is to target EGFR for degradation, which accounts for inhibition of other functions of EGFR apart from its tyrosine kinase activity which are important for cancer cell survival. These elusive functions of EGFR might be governing the activation of compensatory pathway or downstream signaling mediators imparting TKI resistance. Since the majority of NSCLC patients which harbor TKI sensitive mutations also contain T790M mutation, combining the T790M specific third generation inhibitors in combination with first generation inhibitors as a combined therapy could be a preferred therapeutic choice for these tumors. The Holy Grail to maximize the benefit of each NSCLC patient with EGFR activating mutation is to identify the oncogene addiction/s along with EGFR and provide a tailored combination therapy at the beginning of treatment before the resistance develops.
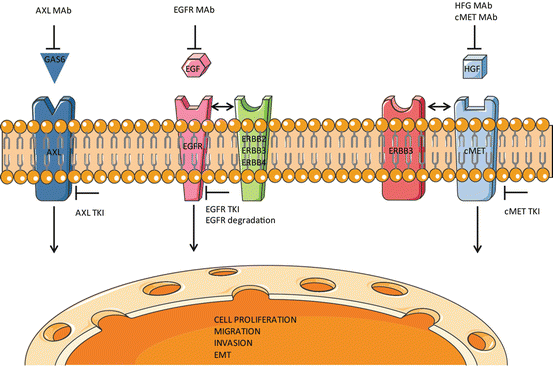
Fig. 1
Major signaling pathways which contribute towards the resistance to EGFR TKIs. In addition to EGFR family members, ERBB2, ERBB3 and ERBB4, AXL and cMET kinases have been reported to mediate EGFR TKI resistance in NSCLC patients. These compensatory pathways can be co-targeted by small molecule inhibitors and antibodies in combination with EGFR TKIs to overcome resistance as shown in the figure
References
1.
2.
Horn L, Chen H, Lovly CM et al (2011) DIRECT: DNA-mutation inventory to refine and enhance cancer treatment—a catalogue of clinically relevant somatic mutations in lung cancer. J Clin Oncol 29(suppl; abstr 7575):494s
3.
Greulich H, Chen TH, Feng W et al (2005) Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med 2, e313PubMedCentralCrossRefPubMed
4.
Politi K, Zakowski MF, Fan PD et al (2006) Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev 20:1496–1510PubMedCentralCrossRefPubMed
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


