MYCOPLASMAL AND UREAPLASMAL INFECTIONS
ARI BITNUN AND SUSAN RICHARDSON
Mycoplasmas and ureaplasmas (class Mollicutes) are the smallest known bacteria (1,2). They are adapted to life in humans, animals, insects, and plants and cannot survive free living in nature due to their dependence on host cells for nutrients. Of the more than 200 known species, about 17 have been associated with human mucous membrane colonization. Three—Mycoplasma pneumoniae, Mycoplasma hominis, and Ureaplasma urealyticum/Ureaplasma parvum—account for most reports of central nervous system (CNS) disease in humans. Rare cases of CNS disease due to other species have been reported, most often in association with immunocompromising or other predisposing conditions (3–5).
Mycoplasmas and ureaplasmas have a cell volume less than 5% that of a typical bacillus such as Escherichia coli and one-sixth the number of genes (about 800,000 base pairs for M. pneumoniae vs. 4.6 million base pairs for E. coli) (1,6). It is thought that they evolved from gram-positive eubacteria by a process of degenerative evolution typified by gradual reduction in genome size (6,7). Due to the lack of a cell wall, they are pleomorphic in shape, are highly susceptible to adverse environmental conditions, and are resistant to β-lactam and glycopeptide antibiotics. Because they do not synthesize folic acid, they are also resistant to the sulfonamides. Their small size allows them to pass through 0.45-µm pore filters commonly used to filter-sterilize media. This trait, coupled with challenges in detection, has earned them wide notoriety for contamination of continuous cell lines. The small genome size also translates into less synthetic capacity and the need for specially enriched media for laboratory cultivation (6).
This chapter covers CNS diseases attributable to M. pneumoniae (Mp), M. hominis (Mh), and Ureaplasma species (Uu). Because the presence of infection or disease in sites other than the nevous system can be helpful in diagnosis, these are discussed as well. In the section on Mp, information related to Guillain-Barré syndrome is included due to overlapping pathogenetic mechanisms. The section on diagnosis includes some basic information on laboratory detection and collection of specimens as a resource for interacting with the clinical laboratory.
MYCOPLASMA PNEUMONIAE
Etiology
Mp is primarily an extracellular pathogen of the respiratory tract. It was first isolated in culture from the sputum of a patient with atypical pneumonia in 1944 (8). Initially referred to as the Eaton agent, it was classified as a “pleuropneumonia-like organism” in 1961 and received its current taxonomic name in 1963 (9). Mp is filamentous in structure (Fig. 25.1), measuring 1 to 2 µm in diameter and 0.1 to 0.2 µm in width. It attaches to human epithelial cells primarily by means of a highly specialized organelle that contains a network of adhesion proteins including the P1 and P30 adhesins (1,6,10). Gliding motility plays an important role in its ability to colonize fully differentiated mucus-producing tissues of the respiratory tract (11,12). It is capable of invading and surviving within cells in vitro (13,14), but whether this occurs to a meaningful extent in vivo is not known. Human-to-human transmission requires close contact and occurs by the droplet contact route.

Epidemiology
Respiratory tract infection due to Mp is extremely common; the incidence of pneumonia is highest in school-aged children (15,16). The proportion of pneumonia cases attributable to Mp increases from about 20% in children between 10 and 16 years of age to about 50% in young adults (17–19). Many infections, however, are subclinical, particularly in those younger than 5 years of age (20,21). There is no particular seasonal distribution. Epidemics occur every 3 to 7 years superimposed on low level but constant endemicity (17).
The incidence of neurologic disease attributable to Mp has been estimated at 0.1% or less (22). Among hospitalized patients with serologically confirmed Mp infection, the incidence is higher, between 1% and 10% (23–25). Neurologic complications are most often sporadic, but clusters of cases have been observed during epidemics of respiratory tract disease (26,27). Children account for 50% to 70% of those diagnosed with neurologic complications of Mp (23,28,29). Furthermore, in Europe and North America, Mp is a leading cause of acute encephalitis in children, responsible for 5% to 13% of all cases (23,28,29). There is no apparent sex predilection (29).
Pathogenesis of Neurologic Disease
The pathogenesis of Mp-associated neurologic disease is incompletely understood. Three broad mechanisms have been proposed with varying levels of supportive evidence (30,31): (a) direct invasion of the brain parenchyma; (b) autoimmunity, or other immune-mediated processes; and (c) vascular occlusion. Neurotoxin-mediated CNS disease, as seen with Mycoplasma gallisepticum in turkeys and Mycoplasma neurolyticum in mice (32,33), has not been demonstrated in humans with Mp-associated neurologic disease.
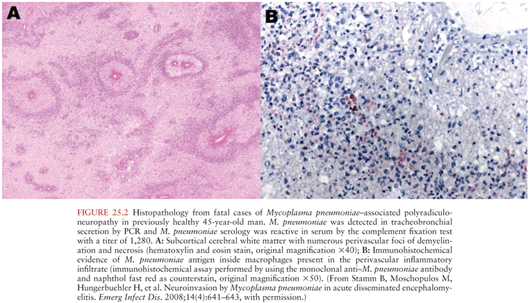
Several lines of evidence support the concept of direct infection of the brain by Mp. Mp has, albeit rarely, been cultured from or detected by various indirect microbiologic techniques at autopsy from the brain of several fatal cases of suspected Mp neurologic disease (Fig. 25.2) (34–37). It has also been cultured from or detected in the cerebrospinal fluid (CSF) by polymerase chain reaction (PCR) of over 50 subjects, 80% of whom had meningitis, meningoencephalitis, or encephalitis (28,38–53). A cerebrovascular vasculopathy characterized by antimycoplasmal immunostaining of endothelial cells and electron microscopic demonstration of Mycoplasma-like structures within endothelial cells has been observed (54,55). Finally, by analogy to other species, there is conclusive evidence from both natural and experimental infection of rodents, turkeys, and alligators by Mycoplasma pulmonis, M. gallisepticum, and Mycoplasma alligatoris, respectively, for the direct infection hypothesis (56–58).
Autoimmunity due to antigenic mimicry and production of antineuronal antibodies has been implicated in the pathogenesis of Mp-associated neurologic syndromes, including postinfectious encephalitis, acute disseminated encephalomyelitis (ADEM), transverse myelitis, and Guillain-Barré syndrome (30,31). Glycolipid epitopes of Mp cross-react with several gangliosides, and anti-galactocerebroside antibodies (anti-GalC) have been demonstrated in the sera of individuals with postinfectious neurologic syndromes due to Mp (59–62). In one study, serum anti-GalC was detected in 3 of 3 individuals with postinfectious CNS disease attributed to Mp as compared to 8 of 32 with Mp infection restricted to the respiratory tract and 2 of 52 healthy controls (61). Among patients enrolled in several Guillain-Barré syndrome treatment trials in Europe, 11 of 16 subjects with anti-GalC had serologic evidence of Mp infection; anti-GalC was present not only in 78% of Mp-associated Guillain-Barré syndrome patients but also in 58% of Mp infections without neurologic disease (63). The antigen mimicry hypothesis is further supported by the observation that the anti-GalC activity in serum of patients with Mp-associated Guillain-Barré syndrome is inhibited by preincubation with Mp antigens (59,60). Among subjects with Mp-associated encephalitis in the California Encephalitis Project, anti-GalC was detected in the CSF of 50% of subjects in whom there was neuroimaging evidence of demyelination (64). Whether anti-GalC causes disease or is an epiphenomenon is uncertain.
With respect to Mp encephalitis, current data suggest that pathogenesis is multifactorial, involving both direct infection and immunologically mediated processes. Several investigators have observed that Mp DNA is detected in serum or CSF of subjects with a prodrome of less than or equal to 7 days but not in those with a longer prodrome (28,48,49). This suggests two distinct patterns of Mp encephalitis: (a) an early-onset syndrome caused by direct invasion and (b) a late-onset syndrome in which the presence of the organism in the CSF or brain is not necessary to cause disease. The CSF cytokine profile of both early- and late-onset forms of Mp encephalitis, consisting of elevated interleukin-6 (IL-6) and IL-8 and normal interferon-γ and tumor necrosis factor-α, suggests a different pathophysiology to that associated with other bacterial and viral pathogens (31,65). The elevated proinflammatory cytokine IL-18 in late-onset, but not early-onset, encephalitis suggests this cytokine may play an important role in the pathogenesis of the late-onset form of disease (31,65).
The pathogenesis of Mp-associated stroke is poorly understood (30,31). A procoagulable state has been implicated in a minority of cases (66–70), whereas in others, a systemic or focal vasculitis appeared to be responsible (71,72). The observation that the mean interval between the onset of respiratory illness and stroke is about 10 days (range 3 to 21 days) is consistent with an immune-mediated process (30).
Clinical Manifestations
Respiratory and Spectrum of Nonneurologic Systemic Manifestations
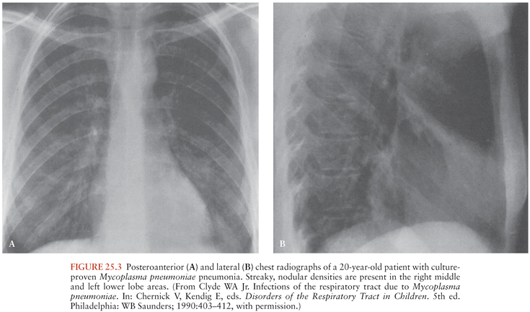
Mp is primarily a pathogen of the respiratory tract. Clinical disease is typified by an insidious onset of fever, headache, malaise, sore throat, and dry cough. In most, disease remains confined to the upper respiratory tract. Progression to tracheobronchitis or pneumonia occurs in fewer than 10% of cases. “Walking pneumonia,” the hallmark of Mp respiratory disease, derives its name from the relatively mild clinical disposition of most of those afflicted. Rales and wheezes may occasionally be heard, but physical examination is often unrevealing despite radiographic evidence of pneumonia. Widespread mottled, diffuse nodular densities are characteristic (Fig. 25.3); pleural effusions occur in 5% to 20% of cases. Although rare, fulminant—and even fatal—disease can occur.
Extrapulmonary manifestations of Mp infection have been described for almost every organ system (1,73). Neurologic complications, followed by those affecting the skin and mucous membranes, are most common. The prototypical dermatologic complication, erythema multiforme minor and Stevens-Johnson syndrome, are seen most often in children and young adults (74–76); in some cases, isolated mucosal involvement can occur (75). Myalgia, arthralgia, and polyarthropathy are relatively common; septic arthritis is restricted primarily to those with immunocompromising conditions (77,78). Cardiac complications include myocarditis, pericarditis, and pericardial effusion. Other extrapulmonary complications include acute glomerulonephritis, tubulointerstitial nephritis, immunoglobulin (Ig) A nephropathy, renal failure, autoimmune hemolytic anemia, thrombocytopenic purpura, and intravascular coagulation (1).
Neurologic Manifestations
The clinical manifestations of Mp-associated neurologic disease are protean and generally indistinguishable from those due to other viral and bacterial pathogens. Syndromes ascribed to Mp, selected key clinical features, strength of the microbiologic evidence supporting the association, and references for each are provided in Table 25.1. It is important to emphasize that for most of the neurologic syndromes associated with Mp infection, proof of causality is lacking. The association is strongest for encephalitis, meningitis, meningoencephalitis, ADEM, transverse myelitis, Guillain-Barré syndrome, and acute striatal necrosis.
A history of respiratory tract infection preceding or accompanying neurologic symptoms is an important clue to the diagnosis of Mp-associated neurologic disease. However, the absence of a respiratory illness does not preclude Mp as a cause; between 35% and 75% of those with Mp-associated acute encephalitis have no history of respiratory symptoms (23,28,29,40,79–81). In those with respiratory symptoms, tracheobronchitis or pneumonia is more typical of adolescents and adults, whereas children younger than 5 years of age often have upper respiratory tract disease characterized by rhinitis, mild cough, or sore throat. In individuals with demyelinating conditions, bilateral striatal necrosis, Bickerstaff brainstem encephalitis, opsoclonus myoclonus syndrome, and stroke, respiratory symptoms are almost universal, preceding neurologic symptom onset by 1 to 4 weeks.
Acute encephalitis is the most common neurologic complication attributable to Mp. Common clinical features include fever (50% to 100%), reduced or altered consciousness (45% to 100%), symptoms or signs of meningeal irritation (20% to 80%), seizures (40% to 60%), focal neurologic deficits (20% to 60%), and ataxia (10% to 25%) (28,40,79). A mild lymphocytic pleocytosis averaging less than 100 cells/µL or a slightly elevated CSF protein is demonstrated in 30% to 60% of cases (28,40,79,81). Nonspecific electroencephalographic (EEG) abnormalities, such as diffuse slowing or findings indicative of an epileptic focus, are observed in 80% to 100% of cases (28,40,79). Periodic lateralizing epileptiform discharges (28) and extreme spindles (82) have been seen rarely. Computed tomography or magnetic resonance imaging abnormalities suggestive of focal edema, ischemia, or inflammation are evident in 35% to 60% of cases (Figs. 25.4 to 25.6) (28,40,81). Mp encephalitis is a severe entity with a mortality of up to 10% (23,79,81) and residual sequelae that include cognitive impairment, seizure disorder, or focal motor deficits in 40% to 60% of survivors (28,79,81,83). In a study of 462 children with encephalitis, those due to Mp were seven times more likely to die or have severe neurologic sequelae than other children in the cohort, second only to herpes simplex virus (84).

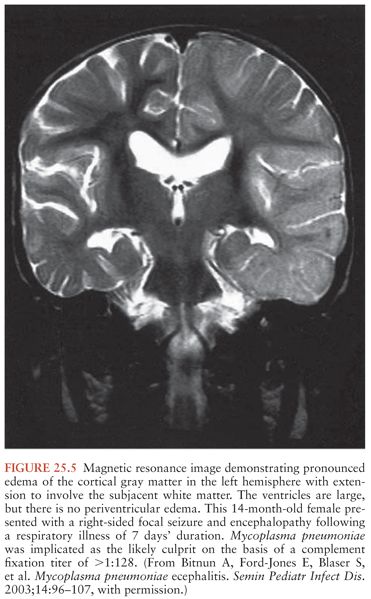
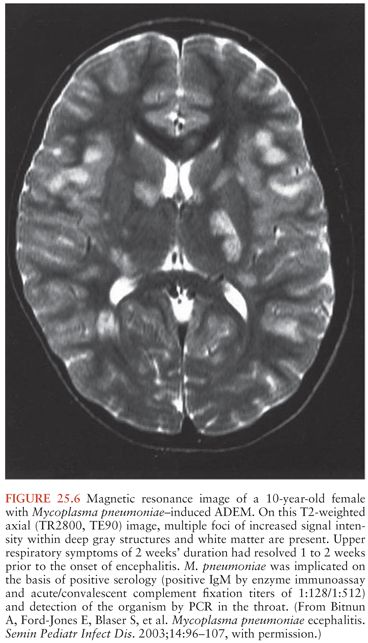
Diagnosis
Mp should be considered in the differential diagnosis of any subject presenting with one of the syndromes associated with Mp regardless of clinical manifestations, CSF profile, and EEG and neuroimaging findings. Current or recent upper or lower respiratory tract infection or pneumonia, whether of undetermined cause or confirmed to be due to Mp, should raise the index of suspicion for Mp as a possible cause. It is worth emphasizing again, however, that the absence of respiratory disease does not preclude the diagnosis, particularly in young children. The presence of nonneurologic extrapulmonary features compatible with Mp may also serve as a clinical clue. The presence of cold hemagglutinins, often used as a quick screen for Mp infection, should not be relied upon due to poor sensitivity and specificity (167).
Microbiologic diagnosis of Mp infection is based on serology and detection of the pathogen by culture or PCR in clinical samples. Culture is of limited use and not routinely available in most clinical laboratories due to its low sensitivity compared to PCR and because it is labor intensive, expensive, and requires up to 3 to 12 weeks of incubation for positive results. The sensitivity of PCR (1 to 10 colony forming units) is 100-fold higher than culture. As Mp neurologic disease can be either due to direct infection of the brain or immunologically mediated, PCR testing or culture of both CSF and respiratory samples is recommended. In one prospective 5-year study, Mp was detected in the CSF of 6 of 11 (54%) children and in the respiratory tract of 5 of 11 (46%) children with probable Mp encephalitis (28). CSF samples intended for Mp PCR should be tested or frozen promptly because the stability of Mp DNA is adversely impacted by storage at room or refrigerator temperatures (168). An important potential limitation of both culture and PCR is that a positive result from a respiratory sample may reflect infection acquired as much as 3 to 7 months earlier and therefore may not be relevant to the acute illness (15,16,169–171). For this reason, combining PCR testing with acute and convalescent serologic testing is encouraged.
Numerous commercial serologic assays are available for the diagnosis of Mp infection (172). Most use a crude culture extract, which contains glycolipid antigens that cross-react with other mycoplasmal, bacterial, human, or plant antigens (173). Better performance (sensitivity and specificity) has been observed when recombinant antigens are used or the antigen is enriched for cytadhesin protein P1, although these assays are generally not commercially available (174,175). In the appropriate clinical context, detection of IgM or IgA in acute sera, seroconversion from negative to positive, or a fourfold rise in titer between acute and convalescent sera is indicative of acute infection (1,6). A negative result, however, does not exclude Mp infection; in a study of 12 commercially available kits in a predominantly adult population with PCR-proven Mp respiratory tract infection, anti-Mp IgM was detected in only 16% to 42% of acute sera and 32% to 84% of convalescent sera (176). Negative serology has also been observed in patients with culture or PCR-proven CNS disease due to Mp (28,34,38).
False-positive serologic results are also a significant concern. For a cohort of children with encephalitis in whom the prevalence of Mp infection is about 7% (28), a false-positive serologic test rate of approximately 50% is expected for an assay with a sensitivity of 90% and a specificity of 94% (30). The observation that 80% of children with encephalitis and reactive Mp IgM in acute sera, in whom the organism was not detected by PCR in either CSF or respiratory samples, had compelling evidence implicating other pathogens as the cause of encephalitis, reinforce this observation and the downside of relying solely on serology for diagnosis (28).
Treatment
The most appropriate therapeutic interventions for suspected or proven CNS disease due to Mp depend largely on the pathogenesis of the syndrome in question. Key considerations are the need for antimicrobial therapy and/or immune-modulating therapies such as corticosteroids, intravenous immune globulin (IVIG), or plasmapheresis (177,178). Because controlled trials evaluating these treatments have not been conducted, recommendations are based on anecdotal evidence from case reports and case series and, in reference to immunologically mediated entities such as ADEM, transverse myelitis, and Guillain-Barré syndrome, extrapolated from data relating to these syndromes irrespective of the infectious trigger. A multidisciplinary care approach is important; for patients with increased intracranial pressure, urgent neurosurgical consultation should be sought as hemicraniectomy may be lifesaving (179).
Antibiotic therapy should be considered for all patients with neurologic disease attributed to Mp, despite the paucity of efficacy data, because of the potential for significant neurologic sequelae. It should be noted, however, that the impact of such therapy on outcome is unknown; full recovery has been observed with (38,39,41,44,51,52,61,83,85,86,97,100,112,155) and without (47,48,61,106) antibiotic therapy, as has an apparent lack of response to such therapy (23,25,52,53,61,83,87,112). In adults and children 8 years of age or older, in whom direct infection of the brain is proven or possible, antibiotics with good in vitro and in vivo activity against Mp capable of traversing the blood–brain barrier and achieving therapeutic levels within the CNS such as azithromycin, doxycycline, or a fluoroquinolone such as moxifloxacin or levofloxacin are preferred (100,180–184). Chloramphenicol is another option as it achieves excellent CSF levels (184) and has good activity against Mp, but the risk of idiosyncratic bone marrow aplasia is of concern. For those with immunologically mediated conditions in whom the CSF tests negative for Mp by PCR, eradication of the pathogen from the respiratory tract using erythromycin or clarithromycin may be appropriate. In children younger than 8 years of age, doxycycline and tetracycline should be avoided due to the risk of enamel hypoplasia and irreversible darkening of permanent teeth.
Immune-modulating therapies are frequently used in the management of demyelinating conditions. Corticosteroids and IVIG, used alone or in combination, have been associated with temporal clinical improvement in some (28,46,51,110–112,114,118–120,185), but not all (36,103,107,186), patients with ADEM and transverse myelitis. IVIG is preferred in Guillain-Barré syndrome (155,156,187). Plasma exchange is reserved for patients (with all three conditions) who fail to respond to corticosteroids or IVIG (103,109,114).
Corticosteroids may also have a role in the treatment of Mp-associated encephalitis. In a case report and retrospective review of severe Mp encephalitis, treatment with corticosteroids was associated with complete or near-complete recovery in 78% of cases (101). By comparison, full recovery was evident in only 52% of children not treated with steroids (79). A prolonged taper over several weeks may be warranted in severe cases to prevent symptomatic relapse (101). The role of immune-modulating therapies in less common forms of Mp neurologic disease is uncertain. Corticosteroids and IVIG, sometimes in combination, have been associated with temporal clinical improvement in several patients with Mp-associated bilateral striatal necrosis (124,135,136) and opsoclonus myoclonus syndrome (145,147,148). IVIG and immune adsorption therapy have been used with apparent success in Bickerstaff brainstem encephalitis (121,122).
OTHER MYCOPLASMA SPECIES
Mh and Uu are rare causes of CNS disease but nevertheless account for the vast majority of mycoplasmal CNS disease not attributable to Mp. Both are commensals of the genitourinary tract. The Ureaplasma genus contains 14 distinct serovars divided into two separate species: U. parvum (serovars 1, 3, 6, and 14) and U. urealyticum (serovars 2, 4, 5, and 7 through 13) (188). It is not known if there are any differences between the two species with respect to pathogenicity, and at present, they can only be differentiated by serotyping or PCR (2,189). For purposes of this review, they are considered together. Rare case reports of CNS disease due to Mycoplasma salivarium, Mycoplasma faucium, Mycoplasma genitalium, and Mycoplasma maculosum have been reported (3–5,190,191). A summary of the salient features of CNS infections due to mycoplasmal species other than Mp is provided in Table 25.2.

MYCOPLASMA HOMINIS
Epidemiology
Mh colonizes the genital tract of 0% to 20% and 10% to 30% of otherwise healthy sexually active men and women, respectively (222–224). Higher rates occur in men with urethritis and in women with bacterial vaginosis, urethritis, or cervicitis (225
Related posts:
 Neurologic Manifestations of Varicella and Herpes Zoster
Neurologic Manifestations of Varicella and Herpes Zoster
 Meningitis and Encephalitis Caused by Mumps Virus
Meningitis and Encephalitis Caused by Mumps Virus
 Human Immunodeficiency Virus
Human Immunodeficiency Virus
 Vaccines for Viral Diseases with Significant Central Nervous System Manifestations
Vaccines for Viral Diseases with Significant Central Nervous System Manifestations
 Subdural Empyema and Suppurative Intracranial Phlebitis
Subdural Empyema and Suppurative Intracranial Phlebitis
 Rickettsioses, Anaplasmoses, and Q Fever
Rickettsioses, Anaplasmoses, and Q Fever
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


