Fig. 19.1
Pie chart showing proportion of colorectal cancer cases believed to be sporadic, familial, or due to hereditary cancer syndromes. HNPCC hereditary nonpolyposis colon cancer, FAP familial adenomatous polyposis
Table 19.1
Genetic syndromes with an inherited predisposition to development of colorectal cancers
Syndrome | Gene defects | Characteristics | CRC risk | Mean age at CRC |
|---|---|---|---|---|
Nonpolyposis syndromes Hereditary nonpolyposis colorectal cancer (HNPCC)/Lynch syndrome | MLH1 (30–40%) MSH2 (50%) MSH6 (7–10%) PMS2 (5%) EPCAM (1–3%) | Autosomal dominant inheritance Synchronous tumors Microsatellite unstable tumors (MSI-H) Other associated cancers : endometrial, ovarian | 50–80% | 40–60 years |
Adenomatous polyposis syndromes Familial adenomatous polyposis (FAP) | APC | Autosomal dominant inheritance 100–1000s adenomatous polyps, duodenal and gastric poylps (fundus) Congenital hypertrophy of retinal pigment epithelium | ≈100% | 30–40 years |
Gardner | APC | Same as FAP—simply a phenotypic variant Additionally associated with desmoid tumors (15%), osteomas of the skull and thyroid cancer | ≈100% | 30–40 years |
Turcot’s | APC (60–70%) | Same as FAP—colon polyps, CRC and CNS tumors primarily medulloblastoma | 50–100% | 20–40 years |
MLH1, PMS (30%) | Lynch type—no polyps, CRC and CNS tumors primarily glioblastoma multiforme | |||
Attenuated adenomatous polyposis coli | APC | Autosomal dominant inheritance <100 polyps Right-sided tumors and polyps with rectal sparing | Unknown | 50 years |
MYH-associated polyposis | MYH | Autosomal recessive inheritance 10–100s adenomatous polyps Somatic KRAS mutation (G12C) Microsatellite stable tumors (MSS) | 80% | 50 years |
Hamartomatous polyposis syndromes Juvenile Polyposis | SMAD4/DPC4 BMPR1a | Autosomal dominant inheritance Hamartoma of colon and stomach, GI bleeding in children <10 years Associated with facial abnormalities including cleft lip or palate and macrocephaly | 60–70% | 60 years |
Peutz–Jeghers | LKB1 STK11 | Autosomal dominant inheritance hamartomatous GI polyps, hyperpigmented macules on the lips and oral mucosa, breast caner | 85% | 50 years |
Cowden | PTEN | Autosomal dominant inheritance Hamartomas of skin, mucus membranes, thyroid, breast | 16% |
Hereditary Nonpolyposis Colorectal Cancer (HNPCC)
Hereditary nonpolyposis colorectal cancer (HNPCC) or Lynch syndrome is the most commonly inherited rectal cancer syndrome and accounts for 2–5% of all rectal cancers (Fig. 19.1). In contrast to other CRC syndromes, these patients do not typically develop adenomatous polyps. This autosomal dominant syndrome results from germline mutations in DNA, mismatch repair (MMR) genes, and accumulation of mutations in microsatellites predisposing to colorectal and endometrial cancers [9–11]. Germline mutations in MSH2 and MLH1 account for 90% of all Lynch syndrome patients, with mutations in MSH6, PMS2, and EPCAM accounting for majority of the rest [12–14] . These patients are also at risk for development of other cancers including ovarian, gastric, and hepatobiliary malignancies. Typically, patients inherit one germline MMR gene mutation and subsequently have an acquired inactivation of remaining allele resulting in frequent mutations and development of microsatellite unstable-high (MSI-H) tumors in the fourth and fifth decade of life. The lifetime risk of colorectal cancer approaches 80%, and so preventative surgery to reduce cancer-related mortality is usually recommended around age 20 years. At the time of diagnosis, approximately 18% of patients will have synchronous tumors, so careful evaluation of the entire colon and rectum prior to surgical intervention must be performed in patients with HNPCC presenting with a cancer [15].
Familial Adenomatous Polyposis (FAP)
FAP accounts for less than 1% of all colorectal cancers. It is an autosomal dominant genetic disorder that results from germline mutations in APC. Phenotypically, FAP manifests as hundreds to thousands of colorectal adenomas beginning in early adolescence. Most patients will have a significant history of early colorectal cancers in first- and second-degree relatives, though about 25% of patients will present as the proband due to “de novo” APC gene mutations [16] . Lifetime incidence of colorectal cancer in patients with FAP approaches 100% with adenomas progressing to invasive cancers by the third or fourth decade of life [17, 18].
Variants of FAP from similar APC gene mutations have also been described and similarly predispose to development of colorectal cancer (Table 19.1).
Patients with FAP also require upper endoscopic surveillance as 90% of patients will develop polyps in the upper GI tract, particularly the duodenum. Duodenal adenocarcinoma is the second most common cause of death in these patients with 5% of duodenal polyps progressing to invasive carcinomas [19, 20]. Attenuated adenomatous polyposis coli is a less aggressive variant of FAP similarly caused by mutations in APC, with a correlation between the severity of the phenotype and the features of the APC mutation. These patients develop fewer polyps that are typically confined to the proximal colon and rarely affect the rectum [21]. Their cancer risk is also lower though the exact incidence is unknown and colon cancers develop later in the fifth decade of life.
Similar to HNPCC, prophylactic colorectal surgery is essential to decrease cancer mortality and must be balanced with quality-of-life measures. Preventative surgery is recommended at age 20 years and must be followed up by frequent surveillance of the remaining GI tract to identify other precancerous polyps.
Other Inherited Rectal Cancer Syndromes
Table 19.1 briefly describes other hereditary syndromes that predispose to the development of rectal cancer. MYH-associated polyposis is an autosomal recessive disorder that results from biallelic germline mutations in MYH, a base excision repair gene. For this reason, patients will not have a generational family history of cancers, but approximately 80% will develop CRC before age 60 years [22]. Other syndromes result in hamartomas of the colon and rectum with varying lifetime risk of development of CRC. SMAD4, PTEN, and STK11 are a few genes implicated in the development of these tumors and also play a role in the development of sporadic rectal cancers as well.
Sporadic Rectal Cancer
The Cancer Genome Atlas (TCGA) Research Network in their comprehensive molecular analysis of colorectal cancer identified two distinct groups or CRC: non-hypermutated and hypermutated (Fig. 19.2a). In their study non-hypermutated tumors accounted for 84% of colorectal cancers and were defined as tumors having somatic mutation rates <8.2 per 106 bases and a median of 58 non-silent mutations [3]. These tumors demonstrated mutations classically described in the early models of colorectal cancer pathogenesis and resulted from somatic copy number alterations resulting in loss of heterozygosity in tumor suppressor genes or oncogene activations. Seventeen genes were noted to be recurrently mutated in this group with mutations in APC, TP53, KRAS, PIK3CA, FBXW7, SMAD4, TCF7L2, and NRAS being most frequent (Fig. 19.2b) [3].
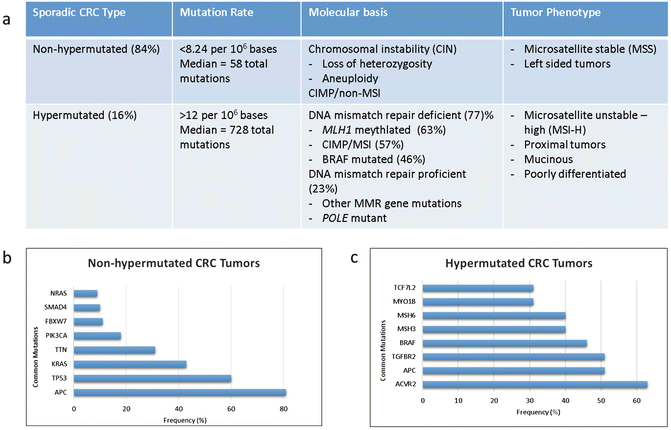
Fig. 19.2
(a) Molecular and clinicopathologic features associated with non-hypermutated and hypermutated colorectal cancer. (b) Genes significantly mutated in non-hypermutated and (c) hypermutated colorectal tumors [23]
Hypermutated tumors were less frequent, accounting for 16% of cases in this study. Hypermutated tumors are underrepresented in rectal cancer population by virtue of their predilection for development in the proximal colon. These tumors driven by deficiencies in DNA damage control have higher rates of mutations (>12 per 106 bases) with a median of 728 total mutations per tumor due to an increase in frameshift mutations [3]. Three quarters of these tumors were microsatellite unstable-high (MSI-H) due to mutations in the DNA mismatch repair pathway, in particular, due to methylation of MLH1 promoter resulting in epigenetic silencing. This commonly occurs in the context of a broader degree of global gene promoter hypermethylation and gene body hypomethylation that is termed the CpG island methylator phenotype (CIMP). Fifteen mutations were frequently mutated including ACVR2A, APC, MSH3, MSH6, and BRAF (Fig. 19.2c). Mutations at DNA polymerase ϵ (POLE) account also result in hypermutated tumors. It is notable that somatic POLE mutations accounted for the highest mutation rates in CRC, but the tumors lack MSI-high, CIMP-high, or MLH1 hypermethylation.
CRC tumorigenesis appears to occur via deferring pathways in these two tumor types. For example, APC and TP53 mutations were common to both groups though at differing rates. These two common drivers were more frequently mutated in non-hypermutated colorectal cancer when compared to hypermutated tumors: 81% vs 51% (p = 0.0023) and 60% vs 20% (p = 0.0001), respectively. Additionally, TP53 loss and mutations in cell cycle checkpoint kinase ATM are mutually exclusive and are found at high frequency in non-hypermutated and hypermutated samples, respectively [23]. And while KRAS mutations are commonly seen across both subsets, BRAF mutations are overrepresented in hypermutated CRC. Additionally, mutations in genes such as TGFBR2 were seen exclusively in hypermutated tumors suggesting that while CRC develops from a known set of deregulated driver pathways, the sequence of genetic events leading to tumorigenesis is different for hypermutated and non-hypermutated CRC.
The CRC Subtyping Consortium (CRCSC) further characterized the genetic and epigenetic features of colorectal cancer by integrating “multi-omic” data regarding tumor mutation status, DNA copy number, methylation, microRNA, and protein expression [24]. In doing so, they identified four distinct colorectal consensus molecular subtypes (CMS), each with their own unique molecular drivers, phenotype, and tumor behavior (Table 19.2). CMS1 (MSI immune) CRC is predominantly comprised of MSI-high tumors. As such, the tumors within this subgroup tend to be hypermutated and hypermethylated and enrich for BRAF mutations and tumor lymphocyte infiltration.
Table 19.2
Consensus molecular subtype (CMS) classification of colorectal cancer. Each subtype reflects unique molecular, genomic, and biological signatures [24]
Subtype | CRC incidence | Driver gene/pathway | Phenotype and prognosis |
CMS1 MSI immune | 14% | Hypermutation MSI-high CIMP-high BRAF mutated Immune activation/expression | Right-side tumors Older age at diagnosis Females Intermediate survival Worse survival after relapse |
CMS2 canonical | 37% | High CIN MSS WNT/MYC pathway activation TP53 mutated EGFR amplification/overexpression | Left-side tumors Better survival |
CMS3 metabolic | 13% | CIN-low Metabolic derangement KRAS mutated PIK3CA mutatedIGFBP2 overexpression | Intermediate survival |
CMS4 mesenchymal | 23% | CIN-high TGF-beta activation NOTCH3/VEGFR2 overexpression | Stromal infiltration Younger age at diagnosisWorse relapse-free and overall survival |
CMS2 (canonical) and CMS3 (metabolic) are driven by chromosomal instability, typically displaying high copy number alterations in microsatellite-stable tumors. CMS2 tumors demonstrate marked upregulation of Wnt and MYC signaling, while CMS4 tumors are characterized by activation of pathways related to mesenchymal transition and stem cell phenotype (e.g., TGF-β, integrins). Additionally, CMS4 tumors have proangiogenic and stromagenic properties that result in stromal infiltration of adjacent cancer tissue [25]. This mesenchymal pathway activation results in early relapse and distant failure in patients with CMS4 mesenchymal CRC even with early-stage disease.
Finally, CMS3 (metabolic) CRC tumors are believed to be driven by metabolic reprogramming with hyperactive glutaminolysis and lipidogenesis that have been shown to contribute significantly to CRC tumorigenesis. CMS3 represents a mixed group with almost one third of tumors being MSI-high, hypermutated, or with intermediate levels of gene hypermethylation. They consistently exhibit fewer copy number alterations and enrich for activating KRAS mutations which have shown to contribute to metabolic adaptation in colorectal cancer and other malignancies [26–31].
While all four of the subtypes can be found in rectal cancer, there is an enrichment for CMS2 and CMS4 compared to proximal colon cancers, where CMS1 and CMS3 tend to be overrepresented.
Targeted Therapy for Rectal Cancer
As previously discussed, dramatic improvements have been seen in rectal cancer overall and disease-free survival with the introduction of cytotoxic chemotherapy agents such as fluorouracil , oxaliplatin, and irinotecan . However, individual responses to these agents vary significantly, and many patients unfortunately suffer the severe side effects of these therapies without deriving any significant benefit. The more precise identification of genetic driver mutations and signaling pathways in colon and rectal cancer have shifted treatment efforts from a population approach to therapy and ushered in a new era of precision medication aimed at personalized treatment regiments that offer patients maximal benefit. These therapies aim to disrupt one or more of the three widely accepted molecular pathways to colorectal tumorigenesis, each contributing to heterogeneity in tumor response and patient survival in rectal cancer (Fig. 19.3).
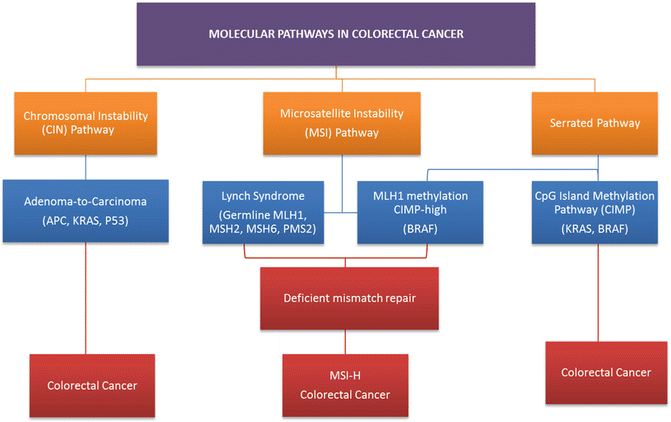
Fig. 19.3
Molecular pathways via which colorectal cancers develop. The chromosomal instability (CIN) pathway accounts for 30–70% of colorectal cancers and results in the progression from adenoma to carcinoma. Germline mismatch repair mutations lead to Lynch syndrome, which accounts for approximately one quarter of all MSI colorectal cancers. The serrated/methylated pathway occurs due to the epigenetic inactivation of MLH1 by hypermethylation resulting in a sporadic MSI phenotype
The classic adenoma-to-carcinoma pathway , also called the chromosomal instability (CIN) pathway , was initially described by Fearon and Vogelstein [32, 33]. They described a multistep progressive model from normal mucosa to adenoma formation and ultimately tumorigenesis characterized by acquired loss-of-function mutations in tumor suppressor genes primarily APC, TP53, and PTEN and mutational activation of oncogenes including RAS and PIK3CA, all leading to chromosomal instability and tumor formation (Fig. 19.4) [33]. Approximately 70–85% of sporadic colorectal cancers arise via this pathway. Over a prolonged period of time, chromosomal rearrangements lead to loss of heterozygosity in tumor suppressors and oncogene activation resulting in accumulation of aneuploidy and deregulated pathways that modulate cellular differentiation, proliferation, and apoptosis. Mutations in the adenomatous polyposis coli (APC) tumor suppressor gene located on chromosome 5q21 typically result in a truncated protein that lacks the ability to appropriately regulate β-catenin degradation [34]. Intracellular accumulation of β-catenin results in deregulation of the Wnt signaling pathway, an important event in tumorigenesis of many solid tumors [35, 36]. APC mutation is believed to be a key player in the adenoma–carcinoma sequence as 40–80% of all CRC tumors carry a mutation in the gene or display allelic losses of 5q [37]. APC mutation events appear to occur early in the adenoma–carcinoma sequence as mutations are seen at a similar frequency in early adenomas when compared to invasive colorectal carcinomas [32, 38, 39]. Genomic studies have shown that pathway alterations in Wnt/β-catenin, transforming growth factor beta (TGF-β), EGFR, and downstream mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) signaling are nearly ubiquitous events in CRC [3, 40, 41]. For this reason, disruption of the Wnt/α-catenin signaling pathway was identified as a potential therapeutic target.
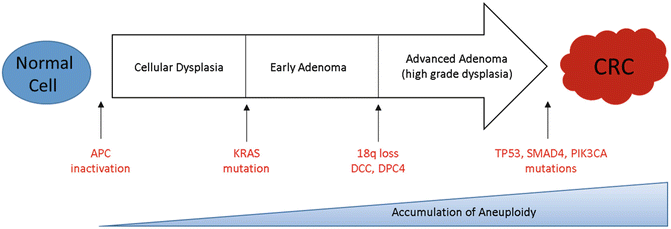
Fig. 19.4
Colorectal cancer tumorigenesis due to the accumulation of genetic alterations annotated with key driver mutations commonly seen in CIN tumors over time
Alternatively, 15% of colorectal tumors can arise from alternate pathways that result in defective DNA mismatch repair due to MLH1, MLH3, MSH2, MSH3, MSH6, or PMS2 inactivation. This impaired repair function results in hypermutation and microsatellite instability (MSI) [23]. This is typified by tumors arising from HNPCC with germline inactivation of mismatch repair proteins and familial MSI-H. A related but distinct pathway, termed the sessile serrated pathway, is typified by early hypermethylation and BRAF mutations with hypermethylation causing silencing of key DNA repair genes, such as MLH1, and leading to sporadic MSI tumors. While typical tubular adenomas share molecular features similar to chromosomally instable CRCs, sessile serrated adenomas arise most commonly in the background of the epigenetic CIMP pathway and MSI which appear to be the major mechanisms driving carcinogenesis [42].
Biomarkers of Prognostic and/or Therapeutic Significance
Vascular Endothelial Growth Factor (VEGF)
Angiogenesis, the recruitment of existing blood vessels and the formation of new capillaries, is vital to tumor growth and establishment. This observation leads researchers to hypothesize that targeting angiogenesis was a viable strategy in cancer therapy [43]. The VEGF family of proteins is comprised of six members, VEGF-A through VEGF-E and placenta growth factor (PIGF)-1 and PIGF-2. VEGF-A, often simply called VEGF, is a potent ligand and regulator of proliferation and angiogenesis in both normal and tumor cells [44]. The VEGF binds to its receptor VEGFR2, a tyrosine kinase, resulting in activation of multiple intracellular signaling pathways and leading to increased endothelial cell proliferation, migration, survival, and ultimately angiogenesis. Not surprisingly, VEGF overexpression has been associated with worse outcomes related to tumor progression and poor overall survival in patients with CRC [45, 46].
Bevacizumab was the first antiangiogenic targeted drug to be approved for the treatment of CRC. Bevacizumab, a recombinant monoclonal antibody against VEGF-A, was heavily anticipated to demonstrate profound antitumoral effect; however, as a single agent the drug showed limited clinical benefit [47]. However, when used in combination with cytotoxic systemic chemotherapies, bevacizumab was shown to increase response rate in patients with mCRC leading to longer PFS and OS [48]. Bevacizumab works synergistically with 5-FU-based chemotherapy to induce apoptosis and inhibit tumor growth presumably by enhancing cytotoxic drug delivery to tumor cells [49]. Consequently, the use of bevacizumab in combination with FOLOX (5-FU, oxaliplatin, leucovorin)/FOLFIRI (5-FU, leucovorin, irinotecan) has been widely employed in the treatment of mCRC, and because of enhanced response rates in the neoadjuvant setting, a significant number of patients with potentially resectable mCRC are later able to undergo curative hepatic metastasectomy. The drug is well tolerated, but potential toxicities can be severe. GI perforation is the most feared and occurs in approximately 1.5% of patients on treatment. Other side effects include hypertension warranting medical therapy, increased risk for stoke and myocardial infarction, and impaired wound healing, though studies have shown that cessation of bevacizumab 4 weeks prior to major surgical intervention poses no increased risk of wound complications [50, 51]. The development of hypertension while on therapy has been associated with a positive treatment response [52, 53].
Epidermal Growth Factor Receptor (EGFR)
EGFR is a tyrosine kinase transmembrane receptor within the HER/ErbB family of proteins [54]. EGFR is overexpressed in tumors of epithelial origin and particularly in colon and rectal cancers. EGFR activation occurs when its primary ligands, epiregulin, amphiregulin, EGF, and TGF-α, bind to initiate activation of multiple signaling pathways. In particular, constitutive activation of the Ras/Raf/MAPK and the phosphatidylinositol 3-kinase (PI3K) signaling pathways lead to deregulated cellular proliferation, inhibited apoptosis, and cellular invasion (Fig. 19.5). EGFR activation therefore contributes not only to tumorigenesis , but is believed to be a key contributor to CRC metastatic progression [55].
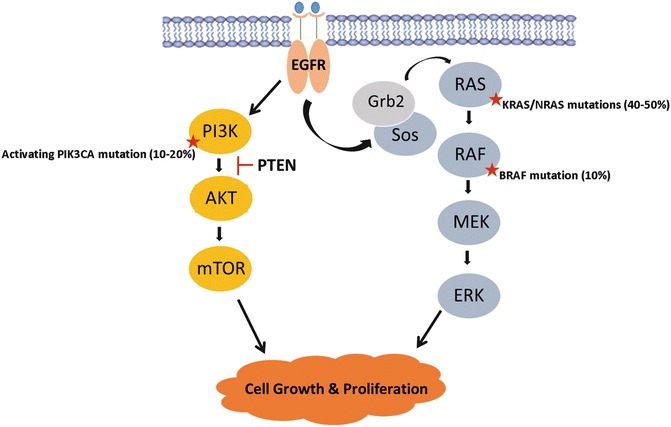
Fig. 19.5
The MAPK signaling pathway is a key player in colorectal cancer tumorigenesis . Mutations in activating RAS, RAF, and PI3K, or mutations in PTEN tumor suppressor, result in constitutive activation of the signaling cascade resulting in uninhibited cellular proliferation and tumor growth
Two classes of anti-EGFR therapies were developed to target this mechanism: tyrosine kinase inhibitors (TKIs) and monoclonal antibodies to EGFR. TKIs, such as gefitinib and erlotinib, competitively inhibit adenosine triphosphate (ATP) to block EGFR activity and preferentially suppress PI3K signaling in CRC [56]. However, efforts to utilize these agents in combination with standard chemotherapy regimens in CRC resulted in high rates of GI toxicity and were not further pursued.
Cetuximab was the first EGFR monoclonal antibody (mAb) to be FDA approved for treatment of metastatic CRC. These antibodies bind EGFR and block ligand binding, thereby inhibiting ligand-dependent autophosphorylation and inhibiting downstream MAPK and PI3K cell signaling. In their seminal paper, Cunningham et al. reported improved response, time to progression, and overall survival with single-agent cetuximab or when used in conjunction with irinotecan in mCRC patients with refractory disease [57]. Similarly, panitumumab was demonstrated to be well tolerated and improved response rates in patients with refractory mCRC and was FDA approved soon after [58]. Anti-EGFR antibody therapy is generally well tolerated. The most common side effect is an acneiform skin rash seen in 80–90% of patients on treatment [59]. Rashes tend to resolve once therapy is discontinued; however, studies have shown a positive correlation in overall survival and severity of skin rash suggesting that this side effect may serve as a forecaster of treatment response [58, 60].
Since their FDA approval in 2004, monoclonal antibodies to EGFR have been widely employed in the treatment of metastatic colon and rectal cancers. However, only few patients derive benefit to therapy with response rates only approaching 10% in most series and prompting efforts to investigate EGFR expression as a prognostic and predictive biomarker [57, 58, 60]. EGFR mutations while common in other tumors are rare in CRC [61, 62]. Early studies explored the association of increased EGFR gene copy or EGFR expression on improved overall survival (OS) and progression-free survival (PFS) in mCRC patients; however, these have not been demonstrated to be reproducibly associated with outcomes and are no longer used for patient selection [60, 63–65]. The investigation of downstream effectors of EGFR signaling, however, has led to the identification of the RAS proto-oncogene family as critical biomarkers in CRC.
KRAS/NRAS
The RAS family of proteins is comprised of three oncogenes (KRAS, NRAS, and HRAS) that code for proteins involved in EGFR-mediated cellular signaling and regulation of proliferation (Fig. 19.5). KRAS mutations occur in 30–40% of colorectal cancers, while NRAS mutations are much less rare with NRAS occurring in 5% of tumors [66–68]. HRAS is not seen in CRC, with mutation rates <0.1%. RAS gene mutations typically occur at codon 12 and less frequently in codons 13, 59, 61, 117, and 146 altering the GTP binding domain dynamics and resulting in constitutive activation of the oncogene. Like CRC, these mutations are observed at high frequency in large adenomas, but less so in smaller adenoma suggesting that they likely contribute more to adenoma growth and CRC carcinogenesis [33, 69, 70]. KRAS and NRAS mutations in colorectal cancers carry both predictive and prognostic significance. Retrospective analysis of several clinical trials of anti-EGFR mAbs revealed that wild-type KRAS was required for anti-EGFR antibody-mediated response in patients with colorectal cancer [71–75]. KRAS/NRAS mutations result in constitutive activation of the Ras-Raf-MAPK pathway, downstream of epidermal growth factor receptor (EGFR), rendering these tumors resistant to anti-EGFR therapies [71, 76–79]. Current guidelines require testing for KRAS and NRAS at the codons above prior to initiating anti-EGFR therapy and established the Ras-Raf-MAPK pathway as a principal target for the development of novel molecular therapeutic agents for the treatment of colorectal cancer [80–82].
While RAS mutation status clearly confers predictive relevance, its prognostic implications require further investigation. While KRAS mutations may be only weakly prognostic in stage II and III colorectal cancer, they may confer poor prognosis in metastatic CRC [83, 84]. Interestingly, exon 146 mutations, though rare, appear to confer better biology than their counterparts with codon 12, 13, or 61 mutations [85].
BRAF
The BRAF oncogene , another potent modulator of the MAPK pathway, has recently emerged as a prognostic biomarker and novel therapeutic target in CRC. BRAF V600E is an activating mutation that accounts for approximately 90% of all BRAF mutations seen in colorectal cancer [79, 86]. As described above, oncogenic BRAF mutations result in tumor formation typically along the serrated pathway by the methylation of CpG islands that cause the silencing of critical tumor suppressor genes and accounting for 10–15% of CRC with discrete clinical characteristics and oncologic outcomes [87–92]. BRAF mutant tumors are more prevalent in women and in patients of advanced age, typically age >70 years [93–96]. Additionally these tumors tend to be proximal in location, MSI-high, of mucinous histology, serrated and poorly differentiated [93–100]. In sporadic colorectal cancers, BRAF mutation is seen in approximately 30–60% of MSI-high tumors but only 5–10% of microsatellite-stable (MSS) tumors [79, 90, 94, 101]. BRAF V600E mutations cause hypermethylation of the MLH1 gene promoter, resulting in loss of the tumor suppressor function and impeding DNA mismatch repair [102–105]. Tran et al. [106] defined BRAF mutant colorectal tumors as a distinct subtype when they delineated a unique pattern of metastatic spread. In their study of a cohort of 524 patients with metastatic colorectal cancer, 11% patients were found to harbor a BRAF mutation. In these patients, metastatic spread was more common via peritoneal disease (46% vs 24%) or distant lymph node metastasis (53% vs 38%) when compared to BRAF wild-type tumors and less likely to result in lung metastasis (35% vs 49%). Not surprisingly, BRAF mutation conferred poorer overall survival with a median of 10.4 months versus 34.7 months. These patients were also less likely to undergo metastasectomy with disease present at sites not amenable to resection. These data implicated BRAF mutation as a major driver of right-sided tumor biology and a potent contributor to poor prognosis observed between right-sided and left-sided colon cancers. Interestingly, the same prognostic implications may not hold for the rare patients with BRAF mutations occurring in rectal primaries, perhaps due to the limited epigenetic alterations seen in these tumors. Further studies are needed to explore this.
Patients with this mutation stand to benefit from effective targeted therapies. The BRAF oncogene codes for a serine/threonine kinase that acts downstream of KRAS in the MAPK pathway (Fig. 19.5). BRAF mediates its effect by activating mitogen-activated protein kinase kinase (MAPKK or MEK) initiating uninhibited EGFR-independent cellular proliferation [90, 107]. BRAF and KRAS/NRAS mutations are mutually exclusive in CRC, suggesting that BRAF is the principal effector of KRAS/NRAS in the MAPK pathway with equivalent effects on tumorigenesis [79, 94, 101]. Consequently, BRAF mutant tumors are able to escape the effects of anti-EGFR therapy similar to RAS mutation and are believed to be responsible for 12%–15% of RAS wild-type patients who fail anti-EGFR therapies [90, 94, 101].
Related posts:
 Histopathologic Evaluation of Neoadjuvant Treatment Response and Tumor Regression Grade
Histopathologic Evaluation of Neoadjuvant Treatment Response and Tumor Regression Grade
 Quality of Life After Multidisciplinary Management of Rectal Cancer
Quality of Life After Multidisciplinary Management of Rectal Cancer
 Palliative and End-of-Life Treatment
Palliative and End-of-Life Treatment
 Combined Transanal/Laparoscopic Total Mesorectal Excision
Combined Transanal/Laparoscopic Total Mesorectal Excision
 Total Mesorectal Excision
Total Mesorectal Excision
 Robotic Rectal Resection
Robotic Rectal Resection
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


