and Infectious Diseases
in Salt Lake City associated with Streptococcus pneumoniae infection. Each pneumococcal strain was determined to be serotype 1, and pulsed-field gel electrophoresis (PFGE) indicated that the isolates were indistinguishable or at least closely related, supporting the hypothesis of clonal spread.2
well. The laboratory techniques used to detect genetic change are also explained in this section before specific molecular typing methods are presented.
typing laboratories. Multilocus enzyme electrophoresis (MLEE) is a protein-based method. With this approach, extracts containing metabolic enzymes from the bacteria are separated by electrophoresis in starch gels. The location for each enzyme in the gel is then detected with a colorimetric substrate specific to that enzyme. Because the electrophoretic mobility of the enzymes depends on their exact amino acid content, it is strain specific. When evaluated as a profile, the electrophoretic mobilities or isoenzyme patterns are referred to as an electrophoretic type. Although it is moderately discriminatory, this technique is not in widespread use because it is technically demanding and has generally been replaced by multi-locus sequence typing (MLST), a method described later in this chapter.15
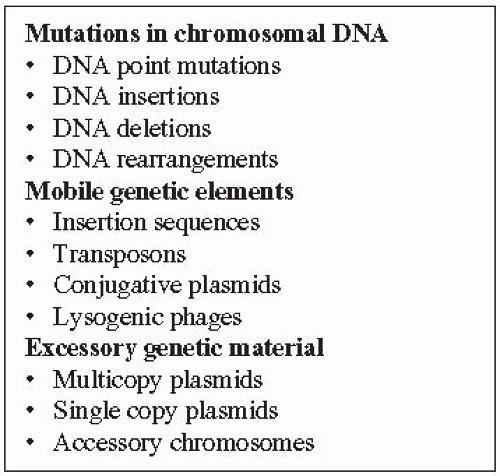 Figure 9-1 Types of Alterations in DNA That Can Be Detected by Molecular Epidemiology |
from a common ancestor. Hence, it is possible to identify numerous changes of subspecies of most bacterial organisms, and to estimate the distance in evolutionary time between isolates.
Table 9-1 Some Common Restriction Endonucleases and Their Recognition Site Specificities | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
If a bacterial species is GC-rich, for instance, a restriction endonuclease whose recognition site is biased toward AT will be an infrequent cutter and a restriction endonuclease whose recognition site has a heavy G+C content will cleave much more often than expected. A good example of this behavior occurs in M. tuberculosis, which is 67% G+C and 33% A+T in its DNA content. The eight-base cutter PacI, which recognizes the AT-rich sequence TTAATTAA, is expected to cut DNA containing equal amounts of AT and GC base pairs about every 65,537 bp. However, because of the heavy G+C content of M. tuberculosis DNA, PacI fails to cut even once within its 4.7 million bp.
Table 9-2 Sequence Recognized by Restriction Enzyme EcoRI | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
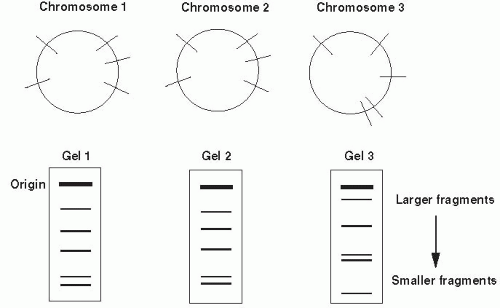 Figure 9-2 Restriction fragment length polymorphisms of three bacterial chromosomes. Lines on circles indicate sites for cutting with a restriction enzyme. Organisms 1 and 2 share restriction endonuclease sites and, therefore, have identical banding patterns on gel electropheresis as depicted. Bacterium 3 has different restriction sites. The fingerprint for organism 3 is different from the other two. |
unrelated and has a different restriction site pattern. The differences between organisms may be visualized by separating the fragments resulting from restriction enzyme digestion by gel electrophoresis. Nucleic acid probes and Southern hybridization can also be used to identify specific restriction fragment differences. Only restriction fragments with specificity for the probe are detected (highlighted bands in Figure 9-2). Southern hybridization using a DNA probe to a region known to be highly variable is an efficient, sensitive way of detecting RFLPs.
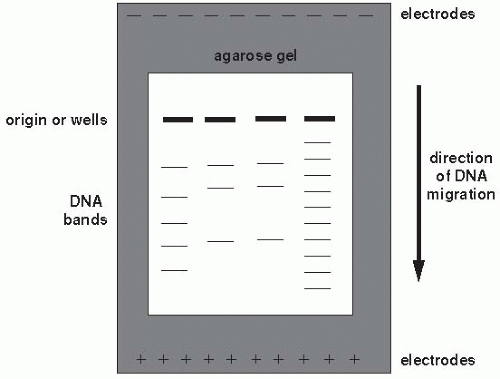 Figure 9-3 Conventional agarose gel electrophoresis. Fourth lane from left indicates molecular weight marker. © 2000, Susan M. Harrington. |
not applied in just one direction as in conventional electrophoresis. Instead, PFGE utilizes alternating electric fields, in which the current is applied for varying lengths of time in each direction, depending on the size of fragments to be separated. Several electrode configurations have been used by investigators; the most popular system is the contour-clamped homogeneous electric field (CHEF). The CHEF apparatus consists of a hexagonal array of electrodes producing two electric fields at 120° angles to each other (Figure 9-4). The length of time that the current is applied in each direction is referred to as the switch time or pulse time. Larger DNA molecules require longer pulse times, whereas smaller fragments are separated adequately with shorter pulse times. PFGE can be used to separate the fragments created by restriction endonuclease digestion of bacterial or fungal genomic DNA. Such digestion generally yields approximately 10 to 20 bands that have a range of fragment sizes. The array of small, medium, and large size fragments is separated by “ramping” the pulse time. With ramping, the pulse time is increased incrementally from just a few seconds up to several minutes over the course of the run.19 Most users of PFGE purchase CHEF equipment, which can perform these intricate electrical switches with little programming by the operator.
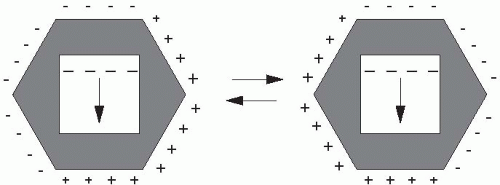 Figure 9-4 Schematic diagram of pulsed-field gel electrophoresis (PFGE) by the contour-clamped homogeneous electric fields (CHEF) technique. Alternation of current is shown. The figure on the left indicates current from northwest to southeast. The figure on the right shows the current from northeast to southwest. © 2000, Susan M. Harrington. |
In a standard PCR reaction, two specific oligonucleotide primers are mixed with template DNA. Oligonucleotides are very short segments of DNA, typically 15 to 30 bp in length. The template is the DNA that contains the sequences to be amplified. Template DNA is generated by lysing bacteria, fungi, or viral particles to release their respective genomes. The oligonucleotides are chosen based on the target sequence to be amplified within the template DNA. One oligonucleotide primer is complementary to the forward (top) strand at one end of the target sequence; the second oligonucleotide is complementary to the reverse (bottom) strand at the opposite end of the target sequence (Figure 9-6).
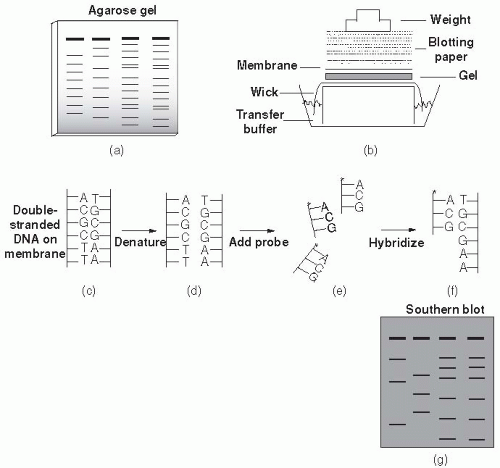 Figure 9-5 (a) Agarose gel electrophoresis. (b) Southern transfer of DNA from agarose gel to nylon membrane, steps involved in hybridization of probe DNA to target DNA on nylon membrane. Double-stranded DNA (c) is separated to the single-stranded form (d). Labeled probe (*) is added (e). Probe hybridizes to complementary DNA to form labeled dsDNA (f). Only bands from the agarose gel (a) with DNA sequence complementary to probe will hybridize. The hybridized Southern blot (g) shows target bands detected by labeled probe. © 2000, Susan M. Harrington. |
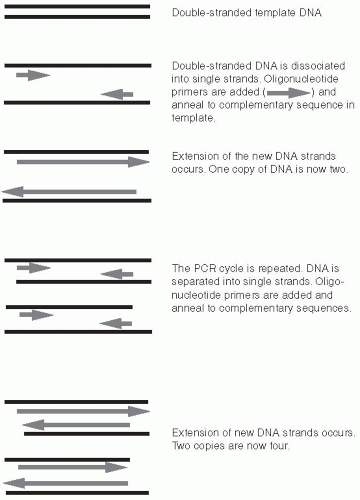 Figure 9-6 Polymerase Chain Reaction Amplification of Template DNA. Two cycles of PCR are shown. Double-stranded DNA is separated into single strands. Primers anneal. New DNA strands are created through extension. Typical PCR reactions are 30 to 40 cycles long, creating millions of copies of double-stranded DNA. |
surface. The location of each DNA on the surface acts as an identifier. Unknown sequences that are complementary to known oligonucleotides on the DNA chip hybridize, allowing for their subsequent identification. Examples of the various formats used for SNP typing with microarrays include hybridization arrays and arrays with enzymatic processing. For hybridization arrays, alleles of known SNPs located at specific regions of a “chip” are allowed to hybridize with query SNPs present in fluorescently labeled PCR products. Hybridization results in positive signals that are detected by a computer, leading to identification of unknown SNPs. During arrayed primer extension, PCR products containing unknown SNPs are hybridized to the arrayed oligonucleotides. Bound oligonucleotides act as primers for a DNA polymerase extension reaction that incorporates fluorescently labeled dideoxynucleotides. The addition of enzymatic discrimination increases the specificity of the latter method.20
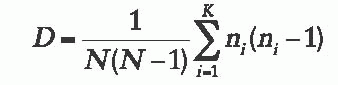
derived by the method, and n is the number of isolates of the ith type. Typing systems with a discriminatory index greater than 0.90 are considered to have effective power.23 One caveat to consider when applying this formula, however, is that the definition of a strain type must be determined; that is, the number of band differences or mutations that must occur for two isolates to be considered different types must be known. Accepted definitions of strain type are defined in the literature for some methods, but not all.
more stable genetic element, not subject to gain and loss as is the plasmid. Chromosomal REA is performed by extracting genomic DNA and cutting it with a restriction endonuclease that cuts frequently. Hundreds of fragments approximately 0.5 to 50 kb in length are produced, which are then separated by conventional gel electrophoresis. The advantages of chromosomal REA are twofold: (1) with the correct selection of restriction enzyme, all bacterial species are typeable, and (2) this technique is easy to perform. However, the large number of bands produced makes interpretation difficult.15, 29 Chromosomal REA has been largely replaced by newer methods.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


