In the last 38 years, a radically new class of infectious agents—the spongiform encephalopathies—has been recognized as the cause of several diseases of humans and animals (Exhibit 21-1). The infectious agents causing these diseases appear to differ from viruses in that they do not contain nucleic acids, but only proteins; they have been labeled prions, or proteinaceous infectious particles by Stanley Prusiner.1, 2 The spongiform encephalopathies are unique among the infectious diseases in that they exhibit none of the traditional hallmarks of an infectious disease. The brain tissue of animals with these infections show spongiform changes in the cytoplasm of nerve cells without an inflammatory infiltrate; the patient or animal does not have a fever, high white cell count, or a rise in acute-phase reactants or cytokines; nor is there a measurable immune response to the infectious agent.3 Therefore, these diseases originally were believed to be degenerative, rather than infectious, and due to genetic factors, rather than infectious agents.
Exhibit 21-1 Transmissible Spongiform Encephalopathies of Animals and Humans (Prion Diseases)
The disease of sheep called scrapie has been known for at least 200 years. It is widespread among sheep in Europe, Asia, and the Americas, and is also seen among goats cohabiting with affected sheep. The Scottish term “scrapie” comes from the characteristic feature of the disease, which is marked by areas of the skin denuded of fleece, due to the animals rubbing their irritated skin against fixed objects. The disease is insidious in onset but is characterized eventually by a progressive, eventually fatal, ataxia that leads to death of the affected animals in a matter of months. The affected sheep are afebrile and have normal cerebrospinal fluid (CSF) and no overt signs of infection. Pathologic lesions are limited to the central nervous system and show the characteristic non-inflammatory spongiform lesions with nerve loss, cytoplasmic vacuolization of degenerating nerves, and a striking astrocytosis. Scrapie initially was believed to be an autosomal dominant genetic disease until two French investigators transmitted the disease to uninfected animals and found the agent to be a filterable agent.4 Subsequently, it was demonstrated that the disease also could be transmitted to mice.5
The scrapie agent is unusual in that it contains no nucleic acids and is resistant to chemicals that normally inactivate nucleic acids, such as formaldehyde, ethanol, ultraviolet (UV) radiation, alkylating agents (such as B-propriolactone), proteases, and nucleases. However, the organism can be inactivated by autoclaving at 121°C under pressure or by exposure to extremes of pH, detergents, or phenol. This led to the hypothesis that the infectious agent was an infectious protein, or prion, which is a proteinase-resistant isoform of a normal cellular protein—that is, it is a normal protein, but is abnormally folded. The prion protein that causes disease has an abnormal structure characterized by a large beta-sheet configuration, in contrast to the alphahelical structure of the normal protease-sensitive host prion protein. It has sometimes been characterized as PrPres or PrPsc compared to PrPc, the normal host prion protein.1, 2
KURU
In the 1950s, a disease was recognized among the primitive Fore people living in a remote area of highland New Guinea. This disease was called Kuru, meaning “shivering” or “trembling” in the Fore language. Kuru is characterized by an insidious onset of truncal ataxia. The ataxia progresses and becomes incapacitating. Eventually, ataxia occurs with every effort of voluntary movement, and death occurs uniformly 3 to 24 months after the onset of symptoms. The pathology shows spongiform changes in the brain similar to that caused by scrapie.
Table 21-1 Transmission of Prion Diseases from Human to Human
Mode of Transmission
Example (no. of cases reported)
Incubation Period (years)
1. Intracranial transplantation or inoculation
a. Dural grafts (>80 cases)
1.3-17
b. Inadequately sterilized instruments (several cases)
0.6-2.2 1.3-1.8
2. Extracranial transplantation
Corneal grafts (2 cases)
1.3-1.5
3. Extracranial inoculation of neural tissue
a. Human growth hormone and gonadotropin (>100 cases)
Because of the ability to transmit scrapie to laboratory animals, William Hadlow, a veterinarian who had worked with the scrapie agent, suggested that Kuru might also be due to a transmissible agent.6 Subsequently, brain tissue from Kuru patients was inoculated into chimpanzees, which resulted in disease.7 Epidemiologic studies subsequently indicated the likelihood that Kuru was due to ritual postmortem cannibalism by the relatives of patients who died from Kuru. While men participated in the cannibalism, Fore adult women and children prepared the body for consumption (see Table 21-1).7, 8 Consequently, the disease was more common in adult women, but male and female children were affected equally, because the women fed infectious material to their children, regardless of their sex.
With the suppression of cannibalism by Australian missionaries and settlers in the 1950s, Kuru has virtually disappeared. There were 11 persons diagnosed with Kuru between 1996 and 2004, indicating an incubation period of more than 50 years.9
CREUTZFELDT-JAKOB DISEASE
Creutzfeldt-Jakob disease (CJD) presents as a dementia, characterized by rapidly progressive mental deterioration, myoclonic jerking, and other neurologic signs. Classic CJD commonly begins between 55 and 70 years of age, but rare cases have occurred in younger adults, even in adolescents. The disease often begins with fatigue, insomnia, and other nonspecific signs or sometimes with focal signs, such as ataxia, visual loss, or aphasia. These symptoms are followed by progressive and relentless dementia, myoclonus, and other neurologic signs. The mean duration of survival is only 5 to 6 months, and more than 80% of patients die within 12 months. As in the other spongiform encephalopathies, there is no fever or other signs of infection, the patient’s spinal fluid is normal, and the brain has characteristic spongiform changes. However, the 14-3-3 brain protein has been reported to be present in the spinal fluid in nearly all patients with CJD.
Approximately 10% of patients with CJD have a family history consistent with an autosomal dominant inheritance. In most, but not all, of these affected families, persons with CJD have a point mutation in the gene coding for the prion protein. The majority of CJD cases (90%) have no other affected family members. CJD occurs at a rate of approximately 1 case per million persons per year without evident clustering (aside from the 10% attributable to familial cases), in virtually all populations.
This disease has been inadvertently transmitted by the transplantation of dural and corneal grafts from infected donors. A total of 230 cases have been recognized worldwide since the first case was reported in 1974; 142 of the 228 cases transmitted by dural grafts occurred in Japan. It has been estimated that as many as 20,000 dural grafts were implanted annually in Japan in the decade before the risk of CJD was recognized. The mean incubation period has been 12 years (range 1.3-30 years) for symptoms of CJD after the graft was placed.10 CJD has also been transmitted by the intracerebral use of a stereotactic electrode to control an epileptogenic focus. Six cases of CJD have resulted from transmission by neurosurgical instruments or electrodes.10 The electrode had been used previously in a CJD patient and subsequently was disinfected only by soaking in alcohol; the CJD prion, like the prion causing scrapie, is resistant to disinfection by viru-cidal compounds.
In addition to these cases resulting from implants or invasive neurosurgical procedures 226 iatrogenic cases of CJD have been transmitted by the use of human growth hormone (HGH) prepared from pools of human pituitary tissue obtained from cadavers. The first three cases of CJD due to growth hormone were reported in 1985. Subsequently, 29 cases have occurred in the United States, 65 cases in the United Kingdom, 119 cases in France, and 13 cases in 6 additional countries.10, 11 In France all 119 cases occurred in a cohort of 1170 patients who received HGH during a 20 month period from December 1985 and July 1985, an attack rate of 10.2%. In the United States CJD has not developed in any patient who started treatment after 1977 when a highly selective column chromatography step was introduced into the purification protocol.10 An estimated 2700 patients received HGH prior to 1997, so the attack rate among patients in the U.S. is estimated at 1.1%. The attack rate in the UK has been estimated at 3.6%.10 Fortunately, HGH is now prepared synthetically, rather than by extraction from pools of human pituitary glands, so no further patients should acquire CJD by this route. However, additional clinical cases may still appear from previous exposure, due to the potentially long incubation period of CJD.
Bovine Spongiform Encephalopathy
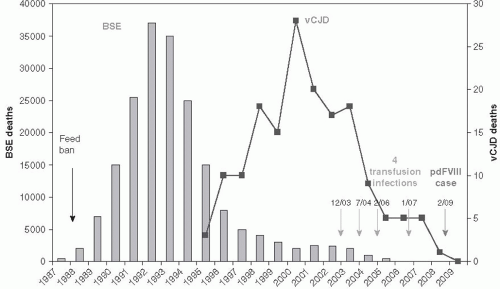
In April 1985, a dairy farmer in the south of England observed a previously healthy cow that became apprehensive, was ataxic, and developed aggressive behavior. Progressive ataxia developed, and eventually the cow died. When tissues were sent to the central veterinary laboratory in the United Kingdom, the brain exhibited the typical features of bovine spongiform encephalopathy (BSE), a disease of cattle known to be caused by infection with a prion. Over the next few years, the number of cases of BSE in cattle in the United Kingdom grew rapidly. Sixteen cases were found in 1986, and more than 7000 cases were found in 1989. The epidemic peaked in 1992, when 36,000 cases of BSE were reported throughout Great Britain, Scotland, and Ireland (Figure 21-1).10
Eventually, more than 220,000 cases of BSE were reported from more than 34,000 herds in the United Kingdom.12 The cattle herds with cases of BSE were scattered throughout the United Kingdom, and there was no evidence of horizontal transmission of the disease between cattle in the same herd. The epidemiologic pattern of disease was that of a common-source, foodborne outbreak. Researchers eventually concluded that the most likely source was from contaminated meat and bone meal (MBM) that had been used for cattle feed. Meat and bone meal had been prepared in the United Kingdom from the rendered carcasses of sheep and other livestock, including cattle. Sheep had long been known to have an endemic level of infection with a prion disease, scrapie, in the United Kingdom. However, BSE did not appear in cattle until the 1980s. On investigation, it was concluded that prions from scrapie-infected sheep and BSE-infected cattle were identical.13, 14
The BSE epidemic followed changes in the cattle rendering process that had occurred in the late 1970s. At that time, changes were introduced into the rendering process for the preparation of bone meal from animal remains (offal), due in part to an oil crisis in the Middle East. The use of continuous heating of offal, as opposed to batch heating, was substituted to save fuel. Also, the sale of tallow became unprofitable because of public concern about the health effects of animal fat consumption. The collapse of the tallow market resulted in most of the rendered fat remaining with the offal; in the past, it had been separated from the offal to be sold. The added fat content probably acted to prevent the disinfection of prion proteins in the animal carcasses. Further amplification of the epidemic was probably related to the use of animal carcasses, including neural tissues from animals such as cattle and sheep that succumbed to BSE or scrapie, for the preparation of animal feed. As a result of these factors, the dose of BSE/scrapie prions in cattle feed probably increased dramatically in the late 1970s.
Bovine spongiform encephalopathy and other prion diseases have been transmitted experimentally to a number of species by the oral route; oral transmission has been accomplished with various species of rodents and nonhuman primates.15 These experimental data supported the hypothesis that BSE could be a foodborne outbreak in the cattle. The theory that the BSE agent in cattle had originated from sheep scrapie was strengthened considerably when molecular genetic studies of the BSE prion protein found it to be identical to the scrapie protein.14
Concern about the growing epidemic of BSE increased until the U.K. government instituted a ban against the use of carcasses from animals at risk for spongiform diseases for the preparation of animal feed. In 1988, a ban on feeding animal-derived feed (such as MBM from rendered sheep, goats, or cattle) to ruminants was instituted in the United Kingdom. This intervention proved to be the critical public health strategy in controlling the BSE epidemic. Nevertheless, as the epidemic evolved, disturbing evidence appeared suggesting cross-species infection from the BSE prion might have occurred. Domestic cats, as well as captive and exotic ruminants, died of BSE after eating animal feed containing possibly infected cattle tissues.16 However, the public health actions that were taken— especially the animal feed ban in 1988—had a dramatic effect on controlling the epidemic after a lag period of approximately 5 years, which is the median incubation period of the disease in cattle. The epidemic peaked in 1992 and has declined progressively since then (Figure 21-1).
The occurrence of BSE in species other than cattle heightened concerns as to whether humans might also be susceptible to transmission of this disease.16, 17 Although no evidence demonstrated that humans were susceptible to scrapie, which had been endemic in sheep in the United Kingdom for two centuries, passage of prions through another animal might have altered the host range. Good evidence related to other prion diseases shows that passage of a prion through one species may alter the host susceptibility in a third species. Experimentally, mouse-adapted strains of scrapie passed through hamsters altered their transmissibility to other rodents,18 human strains of Kuru or CJD could not be transmitted to ferrets until passed through primates or cats,19 and a bovine strain of BSE could not be transmitted to hamsters until it was passed through mice.20 Although the reasons for the species barrier in prion diseases is not known, the theory is that the likelihood of successful interspecies transmission is influenced by the degree of homology between the pathologic prion protein and the host endogenous prion protein.
Variant Creutzfeldt-Jakob Disease
In 1989, specified types of bovine offal (including brain, spinal cord, and organ meats) were banned from human food in the United Kingdom. However, because of concern about the potential transmission of BSE to humans, national surveillance of CJD was instituted in the United Kingdom in 1990. During the interval between 1990 and 1994, no unusual cases or clusters of CJD in humans were encountered.18 Beginning in 1994, however, patients with CJD with an unusual clinical presentation, course, laboratory findings, and brain histopathology were identified. Furthermore, these cases of atypical CJD were much younger than the classic cases. Because of their unusual demographic, clinical, and pathologic features, they were called variant Creutzfeldt-Jakob disease (vCJD).21, 22 The clinical and pathologic differences between classic and variant CJD made it possible to classify patients (Table 21-2). The cases could then be classified definitively by molecular analysis of their prion to determine whether it was a BSE or classic prion protein.
Table 21-2 Comparison of New-Variant and Sporadic Creutzfeldt-Jakob Disease
Characteristic
Variant
Sporadic
Mean age at onset (yr)
29
60
Mean duration of disease (mo)
14
5
Most consistent and prominent early signs
Psychiatric abnormalities, sensory symptoms
Dementia, myoclonus
Cerebellar signs (% of patients)
100
40
Electroencephalographic periodic complexes (% of patients)
 Early History of Infectious Disease: Epidemiology and Control of Infectious Diseases
Early History of Infectious Disease: Epidemiology and Control of Infectious Diseases



