A group of autosomal dominant macrothrombocytopenias caused by mutations in the
MYH9 gene with variable penetrance and expression has been defined.
26,
27,
28,
29 These include May-Hegglin anomaly, Sebastian syndrome, Epstein syndrome, and Fechtner syndrome. These disorders are defined by the presence of mutations involving the
MYH9 gene located at chromosome 22q12.3-13.1 and include single nucleotide changes, in-frame deletions or duplications, and frameshift and nonsense mutations.
30 The
MYH9 gene encodes nonmuscle myosin heavy chain IIA (NMMHC-IIA).
31 NMMHC-IIA is part of the nonmuscle myosin IIA hexamer that is a component of the contractile cytoskeleton in megakaryocytes, platelets, and other tissues. Thrombocytopenia is usually mild and derives from complex defects of megakaryocyte maturation and platelet formation. More specifically, these mutations likely affect the platelet release from the mature megakaryocytes.
30 All of these
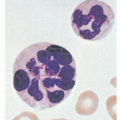
MYH9 gene mutation-related disorders have macrothrombocytopenia, leukocyte inclusion bodies (Döhle body-like inclusions) and can have various combinations of hereditary nephritis, deafness, and cataracts (
Table 49.3). Sometimes, the inclusions are rarely appreciated on conventional May-Grunwald-Giemsa staining, especially in patients with mutations at codon 702 because of their low RNA content. These cases can be diagnosed by the use of immunofluorescence assays.
32 Thrombocytopenia is generally mild to moderate. Peripheral blood smears reveal enlarged platelets with frequent giant platelets.
Clinically, patients have a mild platelet-type bleeding disorder. May-Hegglin anomaly and Sebastian syndrome both have macrothrombocytopenia and granulocyte inclusions, but ultrastructural analysis of the Döhle-like inclusions demonstrates diagnostic differences between these two syndromes, and both types of inclusions can also be distinguished from Döhle bodies seen in acute infection. Patients with Fechtner syndrome and Epstein syndrome have hearing disability and nephritis. In Fechtner syndrome, cataracts are also present.
33 These two disorders result from allelic mutations at amino acid 702, which cause conformational changes to the myosin head.
31,
34,
35,
36 Diagnosis is suggested by detection of macrothrombocytopenia and the presence of one or more of the above-mentioned clinical features. Döhle-like inclusion bodies within neutrophils on May-Grünwald-Giemsa-stained peripheral blood are highly suggestive of an
MYH9-related disorder. Abnormal staining of neutrophil inclusions with anti-NMMHC-IIA may offer greater sensitivity. Definitive diagnosis requires the demonstration of a causative mutation within the
MYH9 gene.
31 Treatment options for bleeding include platelet transfusion, allogeneic stem cell transplantation, or use of a thrombopoietic drug (eltrombopag).
37Platelet-type von Willebrand disease is another autosomal dominant disorder associated with hereditary thrombocytopenia, characterized by abnormal binding of large vWF multimers to platelets. This intrinsic platelet defect results in mild thrombocytopenia, increased ristocetin-induced platelet aggregation, and a selective
loss of high-molecular-weight vWF multimers from the plasma. This disorder resembles type 2b von Willebrand disease (see
Chapter 53).