INTRODUCTION
SUMMARY
Normal hemoglobin can be oxidized to methemoglobin. Methemoglobinemia occurs because of either increased production of oxidized hemoglobin from exposure to environmental agents or diminished reduction of oxidized hemoglobin because of underlying germline mutations. Cyanosis is virtually invariant in patients with methemoglobinemia. Hemoglobin can also bind carbon monoxide and nitric oxide, resulting in the formation of carboxyhemoglobin and nitrosohemoglobin. Sulfhemoglobinemia occurs because of increased production secondary to occupational exposure to sulphur compounds or exposure to oxidant medications. These modified hemoglobins are known as dyshemoglobins. Depending upon the severity and individual predisposition, presence of dyshemoglobins can result in varying degree of clinical manifestations. Prompt diagnosis is the key to effective and timely treatment.
METHEMOGLOBINEMIA
A bluish discoloration of the skin and mucous membrane, designated cyanosis, has been recognized since antiquity as a manifestation of lung or heart disease; however, in methemoglobinemia and sulfhemoglobinemia, it has a different molecular basis than in hemoglobin oxygen desaturation. Cyanosis resulting from drug administration has also been recognized since before 1890.1 Toxic methemoglobinemia occurs when various drugs or toxic substances either oxidize hemoglobin (Hb) directly in the circulation or facilitate its oxidation by molecular oxygen.
In 1912, Sloss and Wybauw2 reported a case of a patient with idiopathic methemoglobinemia. Later Hitzenberger3 suggested that a hereditary form of methemoglobinemia might exist and, subsequently, numerous such cases were reported.4 In 1948, Hörlein and Weber5 described a family in which eight members over four generations had cyanosis. The absorption spectrum of methemoglobin was abnormal and they demonstrated that the defect must reside in the globin portion of the molecule. Subsequently, Singer6 proposed that such abnormal hemoglobins be given the designation hemoglobin M. The cause of another form of methemoglobinemia that occurs independently of drug administration and without the existence of any abnormality of the globin portion of hemoglobin was first explained by Gibson,7 who clearly pointed to the site of the enzyme defect, nicotinamide adenine dinucleotide (reduced form) (NADH) diaphorase, also designated as methemoglobin reductase, and now known as cytochrome b5 reductase. More than 50 years after Gibson’s insightful studies, the genetic disorder that he had predicted was verified at the DNA level.8
Acronyms and Abbreviations:
AOP2, antioxidant protein 2; 2,3-BPG, 2,3-bisphosphoglycerate; cGMP, cyclic guanosine monophosphate; CO, carbon monoxide; COHb, carboxyhemoglobin; GSH, reduced glutathione; N2O3, dinitrogen trioxide; NADH, nicotinamide adenine dinucleotide (reduced form); NADPH, reduced nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; NOS, nitric oxide synthase; P50, the partial pressure of oxygen at which 50 percent of the blood hemoglobin is saturated with oxygen; RBC, red blood cells; SNO-Hb, S-nitrosohemoglobin; SpCO, arterial carboxyhemoglobin concentration; SpMet, arterial methemoglobin concentration; SpO2, arterial oxygen saturation.
The existence of abnormal hemoglobins that cause cyanosis through quite another mechanism was first recognized in 1968 with the description of hemoglobin Kansas.9 Here the cyanosis resulted not from methemoglobin, as occurs in hemoglobin M, but rather from an abnormally low oxygen affinity of the mutant hemoglobin. Thus, at normal oxygen tensions, a large amount of deoxygenated hemoglobin is present in the blood of affected patients.
Methemoglobinemia occurring as a result of cytochrome b5 reductase deficiency is more common among Native Americans, both in Alaska and in the continental United States, and among the Evenk people of Yakutia of Russian Siberia than in other ethnic groups.10,11,12 Methemoglobinemia resulting from hemoglobin M is inherited and sporadic. The occurrence of methemoglobinemia due to toxic chemicals is acquired, transient, and is also sporadic.
Methemoglobinemia decreases the oxygen-carrying capacity of blood because the oxidized iron cannot reversibly bind oxygen. Moreover, when one or more iron atoms have been oxidized, the conformation of hemoglobin is changed so as to increase the oxygen affinity of the remaining ferrous heme groups. In this way methemoglobinemia exerts a dual effect in impairing the supply of oxygen to tissues.13
Hemoglobin is continuously oxidized in vivo from the ferrous to the ferric state. The rate of such oxidation is accelerated by many drugs and toxic chemicals, including sulfonamides, lidocaine and other aniline derivatives, and nitrites. A vast number of chemical substances may cause methemoglobinemia.14,15,16 Table 50–1 lists some of the agents that are responsible for clinically significant methemoglobinemia in clinical practice.
The most common offenders include benzocaine and lidocaine.17,18,19 In some cases, the patients have been unaware that they have been ingesting one of the drugs known to produce methemoglobinemia; dapsone is apparently used in some “street drugs.”20,21 Nitrates and the nitrites contaminating water supplies or used as preservatives in foods are also common offending agents.22,23,24,25,26,27,28,29,30
Cytochrome b5 reductase, also known as NADH diaphorase, catalyzes a step in the major pathway for methemoglobin reduction. This enzyme reduces cytochrome b5, using NADH as a hydrogen donor. The reduced cytochrome b5 reduces, in turn, methemoglobin to hemoglobin. A steady-state methemoglobin level is achieved when the rate of methemoglobin formation equals the rate of methemoglobin reduction either through the cytochrome b5 reductase or through a relatively minor auxiliary mechanism such as direct chemical reduction by ascorbate and reduced glutathione. A reduced nicotinamide adenine dinucleotide phosphate (NADPH)-linked enzyme, NADPH diaphorase, does not play a role in methemoglobin reduction except when a linking dye such as methylene blue is supplied (see “Therapy, Course, and Prognosis” below). A marked diminution in the activity of cytochrome b5 reductase will result in the accumulation of the brown pigment in circulating erythrocytes.
A balance to methemoglobin formation is antioxidant protein 2 (AOP2), which is present in high concentrations in human and mouse red cells (Chap. 47). This member of the peroxiredoxin protein family binds to hemoglobin and prevents both spontaneous and oxidant-induced methemoglobin formation.31 Mutations of this gene or its acquired deficiency are theoretical candidates responsible for congenital and acquired methemoglobinemia. Cyanosis resulting from abnormal hemoglobins (both hemoglobin M and low-oxygen affinity hemoglobins) is inherited as an autosomal dominant disorder. In contrast, hereditary methemoglobinemia resulting from cytochrome b5 reductase deficiency is inherited in an autosomal recessive fashion.
Many mutations of cytochrome b5 reductase that cause methemoglobinemia have been identified at the nucleotide level,8 and the functional effect of some of these have been deduced from the structure of the enzyme.32,33 Although most of the mutants have been found in persons of European descent, five unique mutations were found in Chinese,34 at least three in Thais,35 two in Americans of African descent,36 and one in an Asian Indian.37 In addition, a common polymorphism (allele frequency = 0.023) has been identified in Americans of African descent; it does not appear to impair the activity of the enzyme.38 Most of the patients with cytochrome b5 reductase deficiency merely have methemoglobinemia and the enzyme deficiency is limited to the red cells, and these have been classified as having type I disease. In type II cytochrome b5 reductase deficiency, which represents 10 to 15 percent of cases of enzyme deficient congenital methemoglobinemia, cytochrome b5 reductase is decreased in all cells. In addition to cyanosis, severe developmental abnormalities can occur; most affected infants die in the first year of life.39,40 Patients with this form of disease are afflicted, in addition to methemoglobinemia, with a progressive encephalopathy and mental retardation. The finding that fatty acid elongation is defective in the platelets and leukocytes of such patients41 provides a clue to the type of defect that could occur in the central nervous system, where fatty acid elongation plays an important role in myelination. Rare patients with deficiency of cytochrome b5 reductase in nonerythroid cells do not suffer any neurologic disorder, and it has been suggested that they be designated as having type III disease42; however, existence of such an entity has been challenged and type III disease likely does not exist.43
Heterozygotes for cytochrome b5 reductase deficiency are not usually clinically methemoglobinemic or cyanotic. However, under the stress of administration of drugs that normally induce only slight, clinically unimportant, methemoglobinemia, such persons have been reported to become severely cyanotic because of methemoglobinemia.44 Although in this report the affected patients were Ashkenazi Jews, the prevalence of cytochrome b5 reductase deficiency in 500 unselected Jewish subjects was found to be low.45 In addition, predisposition to acute toxic methemoglobinemia in heterozygous subjects for cytochrome b5 reductase deficiency seems to be quite uncommon.43
Animal models of cytochrome b5 reductase deficiency have been described in dogs, cats, and horses.46,47
A combination of both increased hemoglobin oxidation and decreased methemoglobin reduction also may occur. Because the activity of cytochrome b5 reductase is normally low in newborn infants,48 they are particularly susceptible to the development of methemoglobinemia. Thus, serious degrees of methemoglobinemia have been observed in infants as a result of toxic materials, such as aniline dyes used on diapers,49 and the ingestion of nitrate-contaminated water24,30 and even of beets.50 Bacterial action in the intestinal tract may reduce nitrates to nitrites, which, in turn, cause methemoglobinemia. In rural areas, fatal methemoglobinuria in infants caused by drinking water from wells contaminated with nitrates still occurs.51
Inhaled nitric oxide (NO) is approved for treatment of infants with pulmonary hypertension because of its vasodilatory effect on pulmonary vessels. During the binding and release of NO from hemoglobin, methemoglobin is formed at a higher rate. In one study of 81 premature and 82 term infants, methemoglobin was above 5 percent in preterm infants and between 2.5 and 5 percent in 16 infants.52
Methemoglobinemia occurring in acidotic infants with diarrhea is a syndrome that may have a fatal outcome.53 Such infants have normal red cell cytochrome b5 reductase activity, and the mechanism by which methemoglobinemia occurs is unknown. However, the syndrome seems most common when soy formula is being fed54 and breastfeeding appears to protect against this.51
Rarely, the defect leading to methemoglobinemia may not be in the cytochrome b5 reductase that transfers hydrogen to the cytochrome b5, but rather to a deficiency in the cytochrome b5 itself.55
The molecular mechanisms by which hemoglobin binds oxygen and releases it are discussed in Chap. 49. Heme is held in a hydrophobic “heme pocket” between the E and F α-helices of each of the four globin chains. The iron atom in the heme forms four bonds with the pyrrole nitrogen atoms of the porphyrin ring and a fifth covalent bond with the imidazole nitrogen of a histidine residue in the nearby F α-helix (Fig. 50–1).56 This histidine, residue 87 in the α chain and 92 in the β chain, is designated as the proximal histidine. On the opposite side of the porphyrin ring the iron atom lies adjacent to another histidine residue to which, however, it is not covalently bonded. This distal histidine occupies position 58 in the α chain and position 63 in the β chain. Under normal circumstances oxygen is occasionally discharged from the heme pocket as a superoxide anion, removing an electron from the iron and leaving it in the ferric state. The enzymatic machinery of the red cell efficiently reduces the iron to the divalent form, converting the methemoglobin to hemoglobin (Chap. 47).
Figure 50–1.
Diagrammatic representation of the heme group inserted into the heme pocket. A, Proximal histidine; B, distal histidine. A. In the deoxygenated form the larger ferrous atom lies out of the place of the porphyrin ring. B. In the oxygenated form the now smaller “ferric-like” atom can slip into the plane of the porphyrin ring. As a result, the proximal histidine, and helix F into which it is incorporated, are displaced. (Reproduced with permission from Lehmann H, Huntsman RG: Man’s Haemoglobins. Philadelphia PA: Lippincott Williams & Wilkins; 1974.)
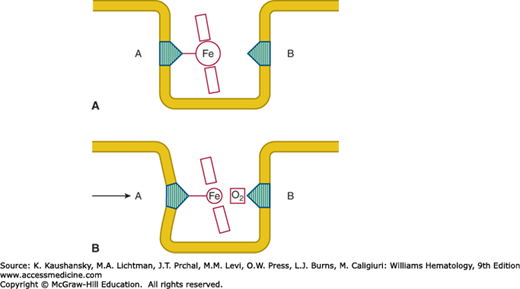
In most of the hemoglobins M, tyrosine has been substituted for either the proximal or the distal histidine. Tyrosine can form an iron–phenolate complex that resists reduction to the divalent state by the normal metabolic systems of the erythrocyte. Four hemoglobins M are a consequence of substitution of tyrosine for histidine in the proximal and distal sites of the α and β chains. As Table 50–2 shows, these four hemoglobins M have been designated by the geographic names of their discovery, Boston, Saskatoon, Iwate, and Hyde Park.
| Hemoglobin | Amino Acid Substitution | Oxygen Dissociation and Other Properties | Clinical Effect | Reference |
|---|---|---|---|---|
| HbMBoston | α58 (E7)His→Tyr | Very low O2 affinity, almost nonexistent heme–heme interaction, no Bohr effect | Cyanosis resulting from formation of methemoglobin | 182 |
| HbMSaskatoon | β63 (E7)His→Tyr | Increased O2 affinity, reduced heme-heme interaction, normal Bohr effect, slightly unstable | Cyanosis resulting from methemoglobin formation, mild hemolytic anemia exacerbated by ingestion of sulfonamides | 182,183 |
| HbMIwate (HbMKankakee, HbMOldenburg, HbMSendai) | α87 (F8)His→Tyr | Low O2 affinity, negligible heme-heme interaction, no Bohr effect | Cyanosis resulting from formation of methemoglobin | 182,184 |
| HbMHyde Park | β92 (F8)His→Tyr | Increased O2 affinity, reduced heme interaction, normal Bohr effect, slightly unstable | Cyanosis resulting from formation of methemoglobin, mild hemolytic anemia | 79 |
| Hb M(hyde park)(HbMilwaukee 2) | ||||
| HbMAkita | ||||
| HbMMilwaukee | β67 (E11)Val →Glu | Low O2 affinity, reduced heme-heme interaction, normal Bohr effect, slightly unstable | Cyanosis resulting from methemoglobin formation | 185 |
| HbFMOsaka | Gγ63His→Tyr | Low O2 affinity, increased Bohr effect, methemoglobinemia | Cyanosis at birth | 57 |
| HbFMFort Ripley | Gγ92His→Tyr | Slightly increased O2 affinity | Cyanosis at birth | 186 |
Analogous His→Tyr substitutions in the γ chain of fetal hemoglobin have also been documented and have been designated hemoglobin FMOsaka57 and FMFort Ripley.58
Another hemoglobin M, HbMMilwaukee, is formed by substitution of glutamic acid for valine in the 67th residue of the β chain. The glutamic acid side chain points toward the heme group and its γ-carboxyl group interacts with the iron atom, stabilizing it in the ferric state.
It is rare for methemoglobinemia to occur as a result of hemoglobinopathies other than hemoglobins M, but HbChile (β28 Leu→Met) is such a hemoglobin. Producing hemolysis only with drug administration, this unstable hemoglobin is characterized clinically by chronic methemoglobinemia.59
Methemoglobinemia may be chronic or acute and acquired or congenital. Acquired severe acute methemoglobinemia, usually the consequence of drug ingestion or toxic exposure, can produce symptoms of anemia, since methemoglobin lacks the capacity to transport oxygen. Symptoms may include shortness of breath, palpitations, and vascular collapse. Chemicals that induce methemoglobinemia are often also capable of causing hemolysis, and a combination of hemolytic anemia and methemoglobinemia may occur. Chronic methemoglobinemia, whether a result of exposure to drugs or toxins or of hereditary causes, is usually asymptomatic. Cyanosis, even if present, may not be discernable in persons with very dark skin coloration.60 In instances when the methemoglobin levels are chronically very high (>20 percent of the total pigment), mild erythrocytosis may be noted (Chap. 57).
Patients with hemoglobin M also manifest cyanosis. In the case of α-globin variants, the dusky color of the infants will be noted at birth, but the clinical manifestations of β-globin variants become apparent only after β chains have largely replaced the fetal γ chains at 6 to 9 months of age. In spite of the impaired hemoglobin function, no cardiopulmonary symptoms are observed and there is no clubbing. In the case of HbMSaskatoon and HbMHyde Park, hemolytic anemia with jaundice may be present. The hemolytic state may be exacerbated by administration of sulfonamides.61
Hereditary methemoglobinemia resulting from cytochrome b5 reductase deficiency may, as noted above, be associated with mental retardation, failure to thrive and early death. In one case, skeletal anomalies were documented as well.62
In toxic methemoglobinemia, an elevated level of methemoglobin is found, but the activity of cytochrome b5 reductase is normal. Methemoglobin levels are best measured using the change of absorbance of methemoglobin at 630 nm that occurs when cyanide is added, converting the methemoglobin to cyanmethemoglobin, a principle used in the Evelyn–Malloy method.63,64 Errors in diagnosis are frequently made when automated instruments designed to estimate levels of reduced hemoglobin, oxygenated hemoglobin, methemoglobin, and carboxyhemoglobin (COHb) are used. Most automated instruments do not properly make this distinction.65,66
The clinical incidence of methemoglobinemia can be overestimated by cooximeter measurements compared to the more specific Evelyn–Malloy method.67 Evelyn-Malloy method involves direct spectrophotometric analysis and should be used when methemoglobinemia is suspected. This is achieved by lysing the blood in a slightly acid buffer and measuring the optical density at 630 nm before and after adding a small amount of neutralized cyanide. The absorption of methemoglobin at this wavelength disappears when it is converted to cyanmethemoglobin. Although this method was described in 1938,63 it remains the most accurate technique for the estimation of methemoglobin in the blood. Details of its performance can be found in an earlier edition of this text68 and elsewhere.61
An eight-wavelength pulse oximeter, Masimo Rad-57 (Rainbow-SET Rad-57 Pulse CO-Oximeter, Masimo Inc, Irvine, CA), has been approved by the FDA for the measurement of both COHb and methemoglobin. The Rad-57 uses eight wavelengths of light instead of the usual two and is thereby able to measure more than two species of human hemoglobin.69 In addition to the usual SpO2 value, the Rad-57 displays SpCO and SpMet, which are the pulse oximeter’s estimates of COHb and methemoglobin concentrations, respectively. In a study on healthy human volunteers in whom controlled levels of methemoglobin and COHb were induced, the Rad-57 measured COHb with an uncertainty of ±2 percent within the range of 0 to 15 percent and measured methemoglobin with an uncertainty of 0.5 percent within the range of 0 to 12 percent,69 the usefulness of this instrument has been verified also by other studies.70,71
In hereditary methemoglobinemia resulting from cytochrome b5 reductase deficiency, between 8 and 40 percent of the hemoglobin is in the form of methemoglobin. The blood may have a chocolate-brown color and cyanosis is present. Cytochrome b5 reductase activity is best measured using ferricyanide as a receptor, measuring the rate of oxidation of NADH.72,73 The residual level of enzyme activity is usually less than 20 percent of normal in patients with methemoglobinemia resulting from deficiency of this enzyme. An immunoassay has been described,74 but such an assay would not detect mutants in which enzyme molecules with impaired catalytic activity are present. For unknown reasons, glutathione reductase activity (Chap. 47) is usually also diminished.75
Cytochrome b5 assays may be useful if cytochrome b5 reductase activity is normal, and the presence of hemoglobin M is ruled out.76
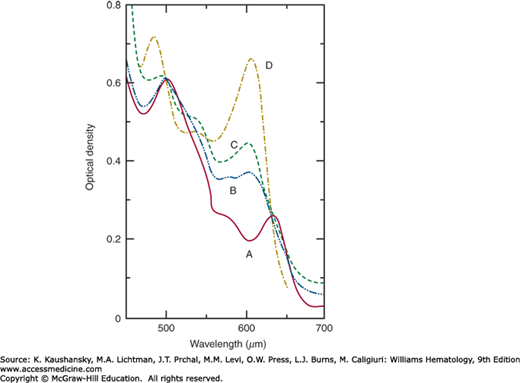
Figure 50-2 illustrates the spectrum of normal methemoglobin A at pH 7.0.77 Hemoglobins M may be differentiated from methemoglobin formed from hemoglobin A by its absorption spectrum in the range of 450 to 750 nm. Because only some 20 to 35 percent of the total hemoglobin will ordinarily be the hemoglobin M, the mixed spectra of methemoglobin A and the hemoglobin M may be difficult to interpret. Therefore, it is preferable to perform these spectral studies on purified hemoglobin M isolated by electrophoretic or chromatographic means.56
Figure 50–2.
Absorption spectra at pH 7.0. A, Methemoglobin A; B, methemoglobin MBoston; C, methemoglobin MSaskatoon; D, methemoglobin A fluoride complex. For purposes of comparison, all the optical densities have been made equal to 0.61 at 500 nm. (Reproduced with permission from Gerald PS, George P: Second spectroscopically abnormal methemoglobin associated with hereditary cyanosis. Science 129(3346):393–394, 1959.)
All hemoglobin M samples should be converted to methemoglobin so that any difference found in electrophoresis will be the result of the amino acid substitution and not the different charge of the iron atom. Electrophoresis at pH 7.1 is most useful for separation of hemoglobins M because the imidazole groups of histidine have a net positive charge at this pH, while at higher pH levels the histidines and the substituting tyrosines are both neutral.
The hemoglobins M differ in their reactivity to cyanide and to azide ions.78 This property may help to identify the subunit affected, as the iron-phenolate bonds are stronger in the α-chain variants than in the β-chain variants. However, definitive identification of the variant requires peptide or DNA analysis. Hemoglobins that cause cyanosis because of a diminished oxygen affinity may be detected by determining the oxygen dissociation curve of blood, being certain that the 2,3-bisphosphoglycerate (2,3-BPG) level is normal, or by estimating the oxygen dissociation curve of hemoglobin, which has been stripped of 2,3-BPG by extensive dialysis against an appropriate buffer. Many of the hemoglobins with decreased oxygen affinity are unstable (Chap. 49) and will precipitate in the isopropanol stability test.78 In many laboratories, it may be easier to analyze the coding sequence of the globin chains at the DNA level than to attempt to determine the properties of the hemoglobin.79
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


