This article gives an overview of meningiomas, pituitary tumors, and intracranial nerve sheath tumors as regards epidemiology, diagnosis, and treatment. Discussion includes the definition of these tumors and their symptomatology, diagnostic procedures, treatment options, surgical techniques, and outcomes.
- •
Microsurgical removal remains the mainstay of therapy for meningiomas and intracranial schwannomas.
- •
If risk of morbidity is increased through, for example, skull base infiltration, multimorbidity, or age, subtotal resection and/or adjuvant radiosurgery is a valuable option.
- •
Even today there is no standard systemic therapeutic option for recurrent tumors, but individual applied therapy regimes are promising.
- •
For nonfunctioning pituitary adenomas, acromegaly, and Cushing disease, transsphenoidal surgery is the treatment of first choice.
- •
Today both microscopic and endoscopic techniques are well established in transnasal surgery. Extended transsphenoidal approaches are increasingly applied in pituitary surgery.
Meningiomas
Biology
Meningiomas represent the most important group of intracranial mesodermal tumors. Harvey Cushing coined the term meningioma in 1922, and it is still the commonly used medical designation for a tumor arising from the meninges, despite the fact that meningiomas actually arise from arachnoid epithelial cells, which are a highly metabolically active subgroup of arachnoid cells involved in the reabsorption of cerebrospinal fluid. However, meningiomas almost always grow in the meninges or, more rarely, in the overlying bone, which they may destroy. Thus, in terms of their manifestations, the term meningioma is completely justified. Meningiomas account for 13% to 26% of all intracranial tumors and are very often benign, indolent, and slow growing, yet quite heterogeneous; as a result their clinical pattern, spontaneous course, and treatment options all differ for each individual case. About 6 of every 100,000 individuals present with a meningioma each year, and 2% of meningiomas are diagnosed in children and adolescents. Peak incidence occurs in the fifth to the seventh decades of life. Women are 2 to 3 times more likely than men to develop a meningioma, whereas malignant meningiomas occur more frequently in men and children. The incidence has increased in recent decades, primarily as a result of increased imaging procedures and rising life expectancy.
Ionizing radiation is a proven risk factor for the occurrence of meningiomas, even at low dose (eg, 8 Gy for the treatment of tinea capitis in Israel during the 1950s), with a latency period of 35 years, and radiation often causes atypical meningiomas with high proliferation indices. Although meningiomas very often express progesterone and estrogen receptors and women develop meningiomas 3 times more frequently than men, and the coincidental occurrence of breast cancer and meningiomas has been accepted, no connection has been demonstrated to date, beyond basic research, between the occurrence of meningiomas and hormonal stimulation. No proof of more frequent occurrence of meningiomas related to increased use of mobile communication devices or as late sequelae of head trauma has been shown in large cross-sectional epidemiologic studies.
The subtype classification established in 2007 includes 9 types of low-grade meningiomas (meningothelial, fibrous, transitional, psammomatous, angiomatous, microcytic, secretory, metaplastic, and lymphoplastic), 3 atypical World Health Organization class II (WHO-II) subtypes (atypical, clear-cell, and chordoid), and 3 malignant types of meningiomas (rhabdoid, papillary, and anaplastic). Atypical and anaplastic meningiomas are more frequently found in men, in whom anaplastic meningiomas occur more often as primary growths, and atypical meningiomas are frequently a manifestation of recurrence in WHO-I tumors. Grade III meningiomas represent about 1% to 3% of all meningiomas, characterized by their high rates of mitosis and nuclear polymorphism. These tumors often infiltrate the brain parenchyma and show signs of tumor necrosis. Typical genetic aberrations found in meningiomas are monosomy 22 and mutations in the neurofibromatosis type 2 gene (NF2), which is localized on chromosome 22. The NF2 gene mutation leads to a defect in the gene product, merlin. Loss of alleles at additional sites can contribute to tumor progression, and lead to the development of atypical and anaplastic meningiomas. The occurrence of a 1p deletion is especially associated with an increased risk of relapse, malignant transformation, and tumor progression.
Most meningiomas have a good prognosis, with the 5-year survival rate more than 80% and the 10-year survival rate 74% to 79%. The spontaneous course involves growth for all meningiomas, but in very different bandwidths (tumor doubling time between 0.5 and 100 years). Calcified, larger, and spinal meningiomas tend to grow more slowly, and tumors in younger people grow more rapidly. The combination of negative progesterone receptor status and a high proliferation index (Ki-67) is considered to have adverse prognostic significance and, despite radical resection, is associated with relapse. Because of their tendency to recur, atypical meningiomas have a 5-year survival rate of 57%. Higher recurrence rates are also associated with incomplete resection (here the Simpson classification developed in 1957 is still the standard ) as well as a higher WHO rating, hemangiopericytic histology, larger tumors, higher mitosis and apoptosis rates, and chromosomal aberrations.
Meningiomas are not characterized by any specific symptoms, and because of their slow and indolent pattern of growth may remain asymptomatic for long periods. Seizures are the most frequent clinical manifestation, occurring in 25% to 40% of cases. A meningioma may become clinically apparent through compression, irritation, or invasion of neighboring brain areas, resulting in symptoms that vary according to the localization and size of the tumor; in addition, they may become symptomatic by causing perifocal edema around the tumor. Increased expression of vascular endothelial growth factor (VEGF) leads to enhanced vascularization and abnormal increases in vascular permeability, which result in edema. Especially in small-sized tumors, it is peritumor edema that may be responsible for the clinical symptoms.
Thus, a parasagittal meningioma in the middle fossa may manifest as disturbances in motor function (parasagittal cortical syndrome, contralateral hemiparesis) which, depending on which brain area is compressed, can result in disturbances of speech, focal seizures, or pareses. Loss of the sense of smell and visual disturbances occur as the result of olfactory meningiomas or tuberculum sellae meningiomas, respectively. Depending on their location, meningiomas of the sphenoid wing may surround the internal carotid artery, the optic nerve, or the oculomotor nerve, and thereby cause deterioration of vision. Cavernous sinus meningiomas affect cranial nerves III, IV, and VI. Occipital headaches with disturbances of gait and dizziness may be a manifestation of a meningioma located at the craniocervical junction. Isolated cranial nerve disturbances (loss of hearing, dizziness, double vision, facial paresis) are often the first symptoms of meningiomas located at the base of the skull.
Diagnosis
Today, magnetic resonance imaging (MRI) represents the most sensitive method for diagnosing and interpreting the differential diagnosis of meningiomas, because in comparison with computed tomography (CT) examination it provides significantly better soft-tissue imaging. Modern MRI sequencing includes special applications for specific issues regarding venous drainage (susceptibility-weighted imaging), MR angiography, MR spectroscopy for the differential diagnosis of metastases or intrinsic brain processes, and diffusion tensor image sequencing for the imaging of possible deviations in the course of the pyramidal tract. However, CT examination can provide essential information about bony infiltration of the meningioma, especially in the area of the sphenoid wing. In addition, demonstration of calcifications on CT may affect the treatment strategy. Angiography is reserved for specific situations, and can provide dynamic information about vascular supply and drainage. If there is a strong tumor blush shown on angiography, selective embolization may make subsequent surgery easier and contribute to improved postoperative outcome. Most meningiomas receive their blood supply from meningeal arteries, but collateral supply through pial vessels may be seen in infiltrative meningiomas with perifocal edema. Using modern nuclear medicine examinations such as positron emission tomography combined with MRI, infiltrative zones can be better represented, thereby optimizing preoperative planning as well as volume planning for radiation therapy.
Surgical Therapy
The treatment regimen for meningiomas must be individualized and interdisciplinary ( Fig. 1 ). As recently as 20 years ago, maximally radical surgery was the preferred treatment option for all meningiomas. However, today the preservation of quality of life stands alongside maximizing life expectancy as the main focus of treatment. Therefore, multimodal treatment programs, with design based on localization, tumor characteristics, and various individual factors such as age, comorbidities, and so forth, have proved to be most effective. Most patients with meningiomas can be cured through surgical resection of their tumors. The urgency of intervention depends on size, localization, and clinical presentation. A regimen of “wait-and-scan” can be justified for asymptomatic tumors regardless of their localization and size, to enable estimation of the dynamics of the process. Meningiomas most frequently occur in the cerebral convexities and the falx cerebri. Less often they arise in the frontal base, the region of the sphenoid wing, the cerebellopontine angle and, least often, in the spinal canal. Interventricular and orbital meningiomas are also very rare. The proportion of the most aggressive and largest tumors is greatest in the cerebral convexity and lowest in the spinal canal.
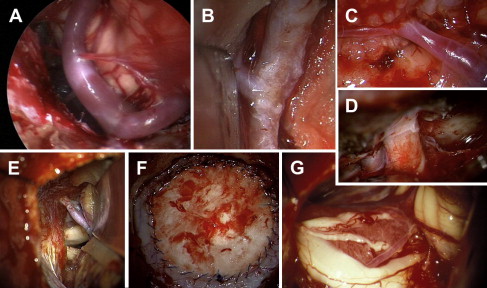
Technical and methodologic innovations in neurosurgery may also be used to provide support in the treatment of meningiomas. Navigational assistance may provide help with minimally invasive procedures and limit the extent of the bony resection required at the base of the skull, while fluorescence-based resections can be helpful in the case of infiltrating recurrent meningiomas and modern ultrasound equipment and neuroendoscopy help visualize the tumor bed.
Because of their surgical accessibility, convexity meningiomas have the highest rate of radical resection. Concomitant resection of the involved dura is critical for preventing recurrence; the question of whether the dural tail demonstrated on MRI represents tumor or reactive dural thickening has not been definitely answered. Infiltration of the venous sinus can be a limiting factor in the extent of the resection. If preoperative imaging (CT angiography, MR angiography) shows a blocked sagittal sinus with the presence of venous collateral circulation, an en bloc resection is possible. Because of the relatively silent brain areas in the frontal cortex, frontobasal meningiomas often remain asymptomatic for long periods of time and are only detected relatively late, when there is loss of the sense of smell or increasing apathy on the part of the patient. Frequently there is transosseous penetration of the tumor through the base of the skull into the sinuses, which necessitates reconstruction of the frontal base after resection of the tumor. Typically it is not possible to preserve the olfactory nerves when treating such tumors. Meningiomas of the cavernous sinus or portions of tumors that infiltrate the cavernous sinus are generally reserved for treatment by radiosurgical procedures, because resection in this area leads to permanent cranial nerve deficits. Maintenance of vital cranial nerve functions is of critical importance in surgery at the base of the skull. Here, intraoperative monitoring plays a key role. Monitoring should include not only surveillance of long tracts (somatosensory-evoked potential, motor-evoked potentials) but also of the cranial nerves in the operative area (N. II–XII), to provide early warning of impending functional disturbances. Spinal meningiomas are a special case. Because of their proximity to the spinal cord, these meningiomas are symptomatic at an early stage and thus are almost always benign (psammomatous), and have a tendency to calcify. Because of their extremely slow growth, even in comparison with intracranial tumors, their size in relation to the spinal cord can be enormous. There is no alternative to surgical resection for spinal meningiomas. Electrophysiologic neuromonitoring for preservation of spinal cord function has proved to be prognostically favorable. Ventral meningiomas growing en plaque cannot be radically resected, and often their dural portion also remains in situ. However, recurrence rates are not any higher, and the Simpson classification does not prove useful here ( Fig. 2 ).
Surgical morbidity rates in the elderly are not significantly increased, and even surgical interventions at the base of the skull can be performed with relative safety. However, careful preoperative assessment is necessary. Serendipitous findings of meningiomas in elderly patients should initially be observed and treated with antiseizure medication, if necessary. Should there be a definite tendency for growth, treatment can be initiated according to the size of the tumor. Radiosurgery is the therapy of choice in patients with multiple morbidities. The treatment regimen for young patients with neurofibromatosis, who often suffer from massive meningiomatosis, should be adapted individually to the patient’s overall clinical status. Radical resections requiring acceptance of a high morbidity risk have just as small a place as primary radiation treatments of meningiomas in very young patients. Because the issue in neurofibromatosis patients is maintenance of quality of life, given their high tumor burden, surgical decompression under continuous electrophysiologic monitoring has proved to be an effective strategy. In the absence of radical treatment, extremely close monitoring of the course of the entire neuroaxis is necessary to detect recurrences.
Radiation Therapy
The focus of therapeutic measures is primarily based on the surgical removal of the tumor, because this is the best way to accomplish immediate reduction in volume. Conventional, fractionated-radiation therapy represents a noninvasive measure primarily used for the subsequent treatment of partially resected tumors. Tumor-conformal irradiation with a sharp fall in the dosage curve at the edge of the tumor, and the low degree of invasiveness of the procedure are critically important. Indications for postoperative radiation are the presence of a malignant or anaplastic meningioma or atypical meningiomas independent of their resection status, and incompletely resected benign meningiomas that show recurrent growth during postoperative follow-up. The recommended total dose is 54 Gy, divided in individual doses of 1.8 Gy 5 times weekly. The target volume includes the tumor (or residual tumor) area plus a safety margin of 2 to 3 cm. For optic sheath and cavernous sinus meningiomas as well as inoperable sphenoid wing meningiomas, fractionated 3-dimensional conformation irradiation is indicated. Malignant meningiomas are irradiated using a total dosage of 60 Gy in 1.8- to 2-Gy individual doses, with the safety margin extended to beyond 2 cm.
Small and well-circumscribed meningiomas with a maximum diameter of 3.5 cm are well suited for stereotactic single-dose radiosurgery using a linear accelerator or Gamma Knife, provided they are located at a sufficient distance from sensitive structures, especially the visual system. The average dose level is about 15 Gy (the minimum dose corresponding to the target volume). Radiosurgery is useful as primary treatment of tumors in a difficult location, in patients with elevated surgical risks, or for meningiomatosis. It is also used as adjuvant treatment after microsurgical partial resection. In individual cases, treatment may be repeated in the event of recurrence or further progression of the tumor.
Chemotherapy
Chemotherapeutic approaches have not shown clear benefit to date. Treatments have been restricted thus far to isolated cases. Experimental approaches using hormone preparations or hydroxyurea (20 mg/kg/d in ongoing treatment over 1 to 2 years) have not yet established a routine place in clinical practice, and their effectiveness still needs to be proved. Antibody-based treatments, including those with platelet-derived growth factor receptor inhibitors (imatinib), somatostatins, temozolomide, and calcium antagonists have not yet been proved as being effective. For anaplastic meningiomas, a therapeutic regimen similar to that used for soft-tissue sarcomas or treatment with an anti-VEGF agent (bevacizumab) may be used as a compassionate intervention.
Despite all of the advances in diagnosis, microsurgical techniques, radiosurgical treatment procedures, and neuropathologic diagnostic methods, many meningiomas continue to present a great challenge that can only be managed through an interdisciplinary approach. There is a persistent conflict between retaining functionality with the aim of a better quality of life, the necessity of treating the tumor, and problems with long-term control of incompletely resected, atypical, or anaplastic meningiomas. These decisions are based on the assessment of the risks and benefits of microsurgical treatment and radiation therapy as well as their long-term consequences, especially regarding the occurrence of secondary tumors and necrosis resulting from radiosurgical procedures. The hope remains that targeted therapy consisting of individualized chemotherapy based on molecular genetic testing of the tumor, will one day be able to resolve the problem of meningiomas that recur despite multiple operations and radiation therapies.
Pituitary tumors
Epidemiology
The prevalence of pituitary adenomas is very high, with 14.4% in autopsy series and 22.5% in radiologic studies. Usually these are asymptomatic microadenomas (<10 mm). A clinically relevant pituitary adenoma requiring treatment occurs in only a small percentage of afflicted patients. A cross-sectional study in the province of Liège (Belgium) revealed 940 clinically relevant pituitary adenomas per 1 million inhabitants. Histologically, pituitary adenomas are usually benign tumors, for which differentiation is made between the frequent, typical pituitary adenomas and the atypical adenomas that show histologic signs of increased proliferation activity. Pituitary carcinomas, defined by cerebrospinal or systemic metastatic spread, are very rare.
Clinically, nonfunctioning pituitary adenomas without hormone secretion are differentiated from functioning pituitary adenomas by the overproduction of a pituitary hormone. A characteristic clinical entity develops in functioning adenomas depending on the pituitary hormone being produced in excess. More than 80% of symptomatic pituitary tumors are pituitary adenomas and a further 10% are craniopharyngiomas. A considerable number of other rare tumor entities are encountered in the pituitary area. This article focuses on the predominant pituitary adenomas.
Clinical Symptoms
Nonfunctioning pituitary adenomas
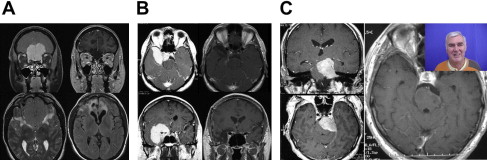
Nonfunctioning pituitary adenomas may be inconspicuous for a long time. Clinical symptoms appear first as a macroadenoma (>10 mm) caused by a local mass effect ( Fig. 3 ). Most often, nonfunctioning pituitary adenomas become clinically manifest with visual impairment (so-called chiasmal syndrome) and symptoms of hypopituitarism. Headache is also often observed as an unspecific symptom. Pituitary apoplexy, caused by a hemorrhagic infarction of a pituitary adenoma, is a dramatic clinical manifestation. Pituitary apoplexy typically leads to acute headache and may be accompanied by nausea, vomiting, meningism, acute visual impairment, pareses of the oculomotor nerves, and high-grade hypopituitarism.
Prolactinoma
Prolactin-producing pituitary adenomas (so-called prolactinomas) are the most common functioning adenomas. Women of child-bearing age are frequently affected. The clinical manifestation is an amenorrhea-galactorrhea syndrome predominantly caused by a microprolactinoma. In men, prolactinoma leads to hypogonadism with impaired libido and sexual potency, as well as infertility.
Acromegaly, gigantism
Growth hormone (GH)-producing adenomas lead to acromegaly with overall increased tissue growth. Externally the typical signs occur, with coarsened facial features, prominent supraorbital ridge, enlarged nose, prognathism, and enlargement of hands and feet; visceromegaly also arises. Other important associated conditions are arterial hypertension, diabetes mellitus, congestive heart failure, arthropathy, goiter, macroglossia, sleep apnea, carpal tunnel syndrome, excessive sweating, and swelling of soft tissue. The increased mortality is mainly due to cardiovascular disease.
Gigantism is the juvenile presentation of a GH-secreting pituitary adenoma. Increased body height results from excessive GH exposure before epiphyseal fusion.
Cushing disease
Corticotropin-producing pituitary adenomas cause excessive cortisol release in the adrenal cortex, which elicits the clinical entity of Cushing disease. The term Cushing disease is used if the cause of hypercortisolism is a pituitary adenoma. Characteristic external stigmata are plethoric moonface, buffalo hump, purple striae, truncal obesity, excessive bruising, and leathery skin. Excess glucocorticoid causes progressive obesity, myopathy, osteoporosis, depression, glucose intolerance, hypercholesterinemia, arterial hypertension, immunosuppression, and hypogonadism. Left untreated, Cushing disease results in death.
Other functioning pituitary adenomas
Other characteristic clinical entities are thyroid-stimulating hormone (TSH)-producing pituitary adenomas and follicle-stimulating hormone (FSH)-producing pituitary adenomas. Thee lesions, however, are much less frequent than the aforementioned entities.
Endocrinologic Diagnostics
The aim of endocrinologic evaluation is to examine pituitary function on the one hand and to prove any hormone oversecretion by the pituitary adenoma.
Anterior pituitary function
Examination of hormones formed in the pituitary gland and of subsequent peripheral hormones is part of basic endocrinologic evaluation. The following hormones are determined in the baseline diagnostics: adrenocorticotropic hormone (corticotropin; ACTH), cortisol, GH, insulin-like growth factor (IGF-1), FSH, luteinizing hormone (LH), estradiol (in women), testosterone (in men), TSH, free triiodothyronine (T3), free thyroxine (T4), and prolactin. Perioperative examination of the adrenal axis is particularly important, because the integrity is essential for a physiologic stress response. If adrenal insufficiency is present, perioperative hydrocortisone replacement is vital. The gold standard for examination of the adrenal axis is the insulin tolerance test. The corticotropin-releasing hormone (CRH) test and the ACTH test are also used as alternatives.
Hormone excess due to functioning adenomas
Prolactinomas
Diagnosis of prolactinomas requires examination of the prolactin level. A 10-fold elevated prolactin level is diagnostic for the presence of a prolactinoma. However, the prolactin level may be lower in the presence of smaller prolactinomas.
Attention must be paid especially to the following in interpreting the prolactin level:
- 1.
The prolactin level can be increased by medication (eg, neuroleptics)
- 2.
Compression of the pituitary stalk by other pituitary tumors leads to a usually mild functional hyperprolactinemia, which should not be confused with the prolactin excess of prolactinomas.
Acromegaly
Two diagnostic criteria are used for proof of acromegaly :
Elevated IGF-1 level as matched for age and gender
Failure to suppress GH in response to an oral glucose tolerance test usually to a level of less than 1 ng/mL; with sensitive GH assays, a threshold of 0.4 ng/mL is suggested.
Cushing disease
The endocrinologic diagnostics of Cushing disease is particularly complicated. First, the presence of a Cushing syndrome must be endocrinologically proven. If the plasma ACTH level is normal or elevated, additional endocrinologic examinations must differentiate between pituitary-dependent Cushing syndrome attributable to an ACTH-producing adenoma (ie, Cushing disease) and ectopic Cushing syndrome. Stimulation of the plasma ACTH and cortisol levels in the CRH-stimulation test and/or suppression of the cortisol level in the high-dose dexamethasone suppression test indicate Cushing disease. If the source of ACTH is unclear, bilateral petrosal sinus sampling is performed. A central/peripheral ACTH gradient greater than 3 after CRH stimulation is proof of pituitary-dependent Cushing syndrome.
Radiologic Diagnostics
The method of choice for neuroradiologic examination is MRI. MRI shows displacement of the pituitary gland and stalk, and the extent of chiasma compression by the pituitary adenoma. About 25% of clinically relevant pituitary adenomas invade the cavernous sinus. Invasive growth can be evaluated using the Knosp criteria.
MRI usually enables a differential-diagnostic categorization of the pituitary tumors. Using MRI, a decision concerning the appropriate surgical approach can be made based on the size and configuration of the tumor. Postoperatively, MRI demonstrates the completeness of surgical resection (see Fig. 3 ).
Ophthalmologic Diagnostics
With suprasellar adenoma extension, the optic chiasm is elevated and compressed, which results in visual dysfunction called chiasmal syndrome with the typical finding of bitemporal hemianopia. Ikeda and Yoshimoto have shown that chiasmal syndrome occurs with a suprasellar displacement of the optic chiasm of more than 8 mm on sagittal MRI.
Thorough ophthalmologic examination is obligatory in all suprasellar pituitary adenomas. The ophthalmologic diagnostics include determination of visual acuity and examination of visual fields. In addition, examination must be made as to whether papillary atrophy is present as an expression of irreversible damage to optic nerve fibers. A complete ophthalmologic status is required to recognize independent ocular diseases.
Operative Treatment
Transsphenoidal operation
More than 90% of all pituitary adenomas can be operated on using a transsphenoidal approach. Since Jules Hardy introduced the surgical microscope in the late 1960s, selective adenomectomy with preservation of the pituitary became possible. Classically the microsurgical technique is performed. In 1987, Griffith and Veerapen described the direct perinasal approach, whereby the preparation begins deep in the nose, after which the bony nasal septum is separated from the rostrum of the sphenoid sinus and displaced to the opposite side using a speculum. The minimally invasive septum-pushover technique, which is based on the technical innovation by Griffith and Veerapen, is widely used today. After the sphenoid sinus is opened and the sellar floor is resected, the adenoma is excised using microinstruments and curettes.
Endoscopy has become established as an alternative operation technique. The purely endoscopic technique was first described by Jho and Carrau, and has continued to gain popularity. Because the optic in the endoscopic technique is placed in the sphenoid sinus, the surgeon has a panoramic overview of the surgical field. The microsurgical technique, on the other hand, provides manual freedom of movement, a 3-dimensional view, and no smearing of the optic system in the case of bleeding, because the microscope is positioned externally. The dispute among experts about which surgical technique is better does not seem justified, because microsurgery and endoscopy differ only in the visualization instrument used.
Extended transnasal approaches
Extended transnasal approaches are increasingly applied in pituitary surgery. The transtuberculum sellae approach enables an intracranial view by removal of the tuberculum sellae. Thus, adenomas attached in the suprasellar space or localized at the pituitary stalk can also be excised. Extended approaches also allow removal of larger adenomas that have developed well beyond the borders of the sella turcica, for example into the cavernosus sinus and the clivus.
Technical support
The use of neuronavigation is very common in neurosurgery today. In transsphenoidal surgery, the authors use neuronavigation in reoperations and in children with nonpneumatized sphenoid sinus as an orientation during the surgical approach. Neuronavigation improves the radicality of the procedure and reduces the surgical risk, because the borders of the tumor and the position of important anatomic structures (eg, the carotid artery) can be identified intraoperatively based on preoperative MRI data. In some neurosurgical centers, such as the authors’ in Tuebingen, intraoperative MRI is available, which allows intraoperative control of the completeness of resection and, if required, further more radical resection during the same surgical procedure.
Results of the transsphenoidal operation
The complication rate of transsphenoidal surgery is low. Typical complications are meningitis and cerebrospinal fluid rhinorrhea. In experienced hands, these 2 complications are in a range of 1% or even less. Life-threatening injury to the carotid artery is very rare. With selective adenomectomy, pituitary function can usually be preserved. New hormone deficits are found in only about 5% of the patients, and a preoperative hypopituitarism may recover at least partially after the operation in up to 50% of the cases. A chiasmal syndrome regresses postoperatively in about 75% of the patients. Postoperative deterioration of vision is rarely encountered. The recurrence rate of nonfunctioning adenomas is low with total resection. However, if adenoma remnants remain, renewed growth must be expected with relatively high probability of recurrence during the further course.
Prolactinomas
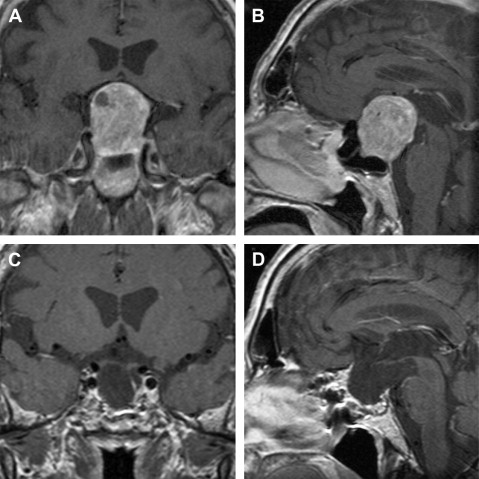
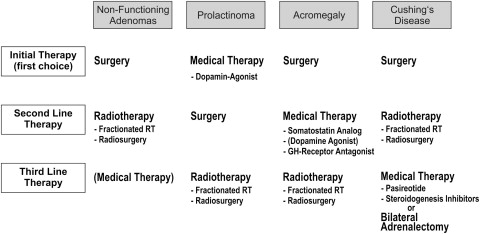
Whereas surgery is the initial therapy of first choice in nonfunctioning adenomas, acromegaly, and Cushing disease, medical therapy is usually the initial treatment in prolactinomas ( Fig. 4 ). However, transsphenoidal surgery has regained recognition in the treatment of prolactinomas in the past years. Long-term remission can be achieved in more than 90% of microprolactinomas, whereas dopamine agonist (DA) therapy must often be continued for life. For this reason, an increasing number of those affected choose the surgical option. The operation is often performed before family planning, because the female patients can then become pregnant without medical treatment. Surgical treatment can also be offered to very young patients and children to avoid the risks of medical therapy over decades. Further indications for the operation are acute visual loss in macroadenomas, nonresponsiveness to DA therapy, or intolerance to DA.
Acromegaly
For complete remission a postoperative GH level of less than 2.5 ng/mL, suppression of growth hormone to less than 1 ng/mL during a glucose tolerance test, and an IGF-1 level within the normal range are required. The largest published operative study described an overall remission rate of 57.3%, but the chance of remission depends in great measure on the size and invasive behavior of the pituitary adenoma. The remission rates for microadenomas were 75%, and 74% for intrasellar macroadenomas.
Regarding Cushing disease, in the literature the initial success rate of transsphenoidal operation is between 70% and 90%. Because of the severity of the disease, the authors favor early reoperation following failure of the first operation.
Transcranial operation
Transcranial operation is indicated if the adenoma configuration suggests low chance for removal of the suprasellar adenoma. This is the case in adenomas with multilobulated suprasellar extension, which indicates a perforation of the diaphragma sellae. A small sella turcica and a narrow sellar entrance may also make craniotomy necessary for the resection of an adenoma with suprasellar growth.
Medical Therapy
Nonfunctioning adenomas
No established medical treatment is available for nonfunctioning adenomas.
Prolactinomas
The prolactinoma is the best example of a tumor that responds to medical treatment (see Fig. 4 ). With the second-generation DAs such as cabergolin, treatment is more effective and has fewer side effects than the classic DA bromocriptine. The excellent results are, however, relativized by a current study, which revealed side effects in 42% of patients during the long-term course. The DA therapy even had to be withdrawn in 18% of patients because of side effects. At present, there are also reservations concerning drug therapy, due to more recent knowledge that long-term administration of DAs may lead to restrictive cardiac valvulopathy.
A meta-analysis revealed a pooled proportion of patients with persisting normoprolactinemia after DA withdrawal of only 21%.
Acromegaly
The most important indication for medical therapy is a postoperative persistence of GH excess (see Fig. 4 ). Somatostatin analogues are the drug-treatment option of first choice. These agents develop their effect via somatostatin receptors directly on the adenoma and, in addition to decreasing GH, may also effect tumor shrinkage. The currently used somatostatin analogues are lanreotide and octreotide. A meta-analysis showed IGF-1 normalization in 47% of the patients under treatment with lanreotide and in 67% of those under treatment with octreotide. DAs can be tried if GH is only slightly increased, but they are less effective in eradicating the GH excess and normalizing IGF-1. Some years ago, the GH-receptor antagonist pegvisomant became available for the treatment of acromegaly. Pegvisomant acts in the periphery and blocks IGF-1 production, and is used if the response to somatostatin analogues is insufficient. Normalization of IGF-1 can be achieved in 62% to 78% of the patients.
Cushing disease
It is difficult to achieve long-term control of hypercortisolism with medical treatment. Medical treatment can be applied for acute treatment of severe Cushing syndrome. Medical therapy is also suitable for patients awaiting a response to radiation therapy and whenever a palliative treatment is needed (see Fig. 4 ). Steroidogenesis inhibitors, which act on the adrenal gland, can be used for medical therapy. The most experience with this drug group has been acquired with metyrapone and ketoconazole.
Other medicaments develop their effect centrally on the adenoma. The somatostatin analogues octreotide and lanreotide have generally been found to be ineffective. Encouraging results have been obtained with the novel somatostatin analogue pasireotide. Pasireotide has a particularly high affinity to the somatostatin receptor subtype 5, which predominates in ACTH-producing pituitary adenomas.
Aggressive pituitary adenomas
Successful treatment of aggressive pituitary adenomas and pituitary carcinomas with the chemotherapeutic temozolomide was recently reported. This result opens a perspective for treatment of functioning and nonfunctioning adenomas that no longer respond to conventional therapy.
Radiotherapy
In nonfunctioning pituitary adenomas, radiotherapy (RT) is usually performed in residual or recurrent adenomas that cannot be surgically removed (see Fig. 4 ). In the case of small residual adenomas, RT can be postponed until there is proof of adenoma progression. In Cushing disease, RT can be used as second-line treatment if surgery fails to cure the patient (see Fig. 4 ). In prolactinomas and acromegaly, RT is the third-line treatment and is indicated when surgical and medical options have been exhausted. Two principal radiotherapeutic techniques are available: fractionated RT and stereotactic singe-fraction radiotherapy (ie, radiosurgery).
Fractionated radiotherapy
Today, conventional fractionated RT is performed with mask-fixing and has been refined to tumor-conformal delivery. Fractionated RT is performed in adenomas in the immediate vicinity of the optic pathway and in large-irradiation volumes. The usual radiation doses are 45 to 54 Gy, applied in fractions of 1.8 (to 2) Gy. The risk of delayed-onset hypopituitarism is more than 50%. Infrequent complications are optic neuropathy, secondary malignoma, and cerebrovascular insults.
In nonfunctioning adenomas, the rate of recurrence-free survival 10 years following surgery and fractionated RT is 80% to 97%. Remission rates for Cushing disease reported in the literature range from 10% to 83%. The UK National Acromegaly Register Study Group reported on 884 acromegalic patients who received conventional pituitary irradiation and found a normalization rate of 63% for IGF-1 at 10 years’ follow-up. However, the withdrawal of suppressive medications is not specified. The remission rates are lower in prolactinomas than in Cushing disease and acromegaly.
Radiosurgery
Single-fraction radiosurgery is particularly indicated for small invasive residual adenomas in the cavernous sinus that are not amenable to surgery. The following modalities are available for radiosurgery: Gamma Knife radiosurgery, linear accelerator (LINAC)-based radiosurgery, Cyberknife radiosurgery, and proton-beam radiation. To prevent optic neuropathy, the dose delivered to the optic apparatus should not exceed 10 to 12 Gy. Advantages of radiosurgery over fractionated RT are the lower rate of hypopituitarism and, in cases of functioning adenomas, the more rapid decrease of the hormone excess, which usually starts within a few months. A comprehensive review of the literature revealed a tumor control rate greater than 90% for nonfunctioning pituitary adenomas with tumor margin doses of 14 to 25 Gy, but the data on long-term follow-up are sparse. Higher radiation doses are required to correct hormone excess than are required to prevent tumor growth. Tumor-margin doses from 15 to 32 Gy are usually reported for functioning adenomas. In large contemporary series, the rates of biochemical remission are approximately 50% for Cushing disease, 35% for acromegaly, and 25% for prolactinoma.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


