purines (reactions 10 and 11), and pyrimidines (reaction 2). By indirectly contributing to S-adenosylmethionine (AdoMet) generation, folate is linked to important methylations, including methylation of DNA cytosine (Fig. 36.2, reaction 13). Cellular folate metabolism and its compartments are reviewed elsewhere.18, 19
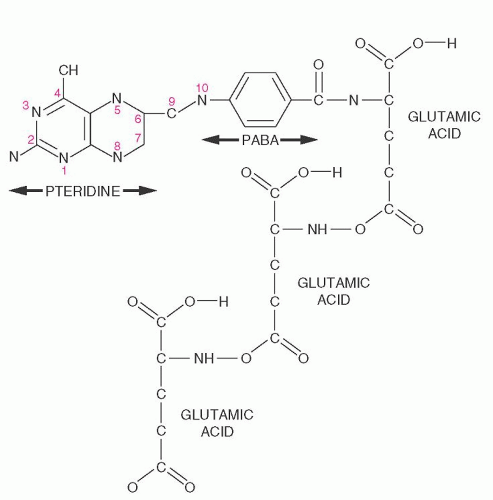 FIGURE 36.1. Folate structure. The constituents are, from left to right, pteridine and p-aminobenzoic acid (PABA), which constitute the pteroyl moiety, and one or more glutamates that are attached by γ-carboxyl linkage (in this diagram, three glutamates are linked). Metabolic activity requires reduction to tetrahydrofolate at positions 5, 6, 7, and 8. Various one-carbon moieties are attached to the nitrogen at position 5 (N5-methyl, N5-formyl, or N5-formimino) or 10 (N10-formyl), or bridging 5 and 10 (N5,10-methylene or N5,10-methenyl). Each folate participates in specific reactions by transferring, accepting, or transforming its one-carbon moiety. |
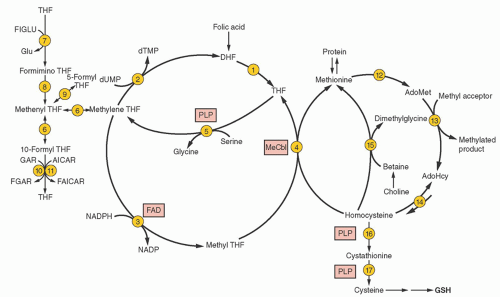 FIGURE 36.2. Folate metabolism and its linkage with cobalamin (reaction 4) to homocysteine and methionine metabolism (right). Vitamin co-factors are highlighted in boxes. Not all bidirectional reactions are noted as such, but the unidirectional nature of reaction 3 is the basis for the methyltetrahydrofolate trap hypothesis. The enzymes for the reactions include the following: (1) dihydrofolate (DHF) reductase; (2) thymidylate synthase; (3) methylene tetrahydrofolate (THF) reductase; (4) methionine synthase (maintained by a reductive system mediated by methionine synthase reductase); (5) serine hydroxymethyltransferase; (6) two sequential reactions mediated by two enzymatic activities of a single protein (methyleneTHF dehydrogenase and methenylTHF cyclohydrolase); (7) glutamate formiminotransferase; (8) formiminoTHF cyclodeaminase; (9) methenylTHF synthase; (10) glycinamide ribonucleotide (GAR) transformylase; (11) 5-aminoimidazole-4carboxamide ribonucleotide (AICAR) transformylase; (12) methionine adenosyltransferase; (13) diverse S-adenosylmethionine (AdoMet)-dependent methyltransferases; (14) S-adenosylhomocysteine (AdoHcy) hydrolase (kinetics usually favor synthesis of AdoHcy); (15) betaine:homocysteine methyltransferase; (16) cystathionine β-synthase;(17) cystathionine γ-lyase. dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; FAD, flavin adenine dinucleotide (riboflavin); FAICAR, formylAICAR; FGAR, formylGAR; FIGLU, formiminoglutamic acid; Glu, glutamic acid; MeCbl, methylcobalamin; PLP, pyridoxal 5′-phosphate. |
by inhibiting both methyleneTHF reductase (reaction 3) and betaine hydroxymethyltransferase (an alternate remethylator of homocysteine [reaction 15] confined to liver and perhaps kidney and lens), and by activating cystathionine β-synthase (reaction 16).29
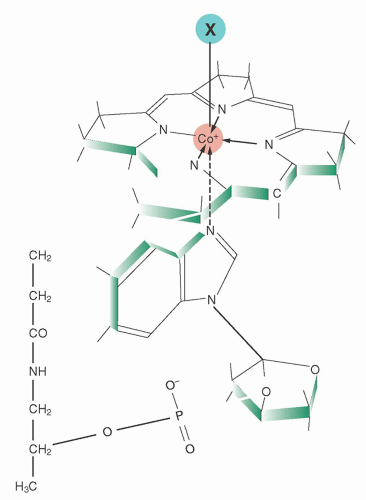 FIGURE 36.3. Cobalamin structure. Attached to the central cobalt atom of the corrin tetrapyrrole and to one of the pyrrole rings is the α-ligand, the 5,6-dimethylbenzimidazole nucleotide, extending below the corrin plane. The β-ligand (marked as X in the diagram) above the plane can be any one of several moieties, such as methyl, 5′-deoxyadenosyl, hydroxyl, or cyanide. |
pH and operating at higher concentrations of folate, reduced folate carrier assumes major responsibility for folate uptake from the blood by the liver, kidney, leukocytes, placenta, and parts of the brain.
Holoprotein proportions can vary widely in some disorders, and the reason is not always known.76, 78 Desialylated TC I is cleared nonspecifically by hepatic receptors for asialoglycoproteins,79 whereby some of its cobalamin is cleared in bile.
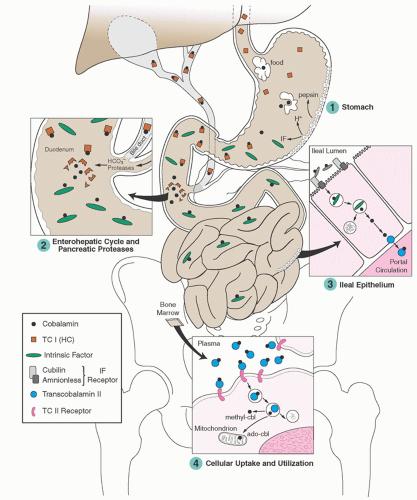 FIGURE 36.4. Assimilation and utilization of cobalamin. ado-cbl, 5′-deoxyadenosylcobalamin; IF, intrinsic factor; methyl-cbl, methylcobalamin; TC I (HC), transcobalamin I (haptocorrin); TC II, transcobalamin II. (Modified from Carmel R. Cobalamin deficiency. In Carmel R, Jacobsen DW, eds. Homocysteine in health and disease. Cambridge, MA: Cambridge University Press, 2001:289-305, with permission.) |
the vast human microbiome in the gut and elsewhere, as well as withholding unusable analogs from human cells.80 The subject requires careful study. Several minor cobalamin-binding proteins and complexes have also been described in plasma, but their roles are unknown.56, 79, 81, 82, 83
TABLE 36.1 COBALAMIN-BINDING PROTEINS | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
associated with broad malnutrition and with alcohol abuse. In contrast, the evolution of cobalamin deficiency is usually measured in years, and it tends to be a purer deficiency state because malabsorption is often restricted to cobalamin alone.
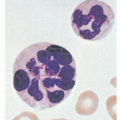
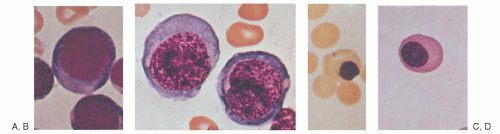 FIGURE 36.5. Normal and megaloblastic precursor cells in the bone marrow. A: Pronormoblast. B: Megaloblastic equivalent of cell in plate A. C: Late normoblast. D: Megaloblastic equivalent of cell in plate C. (From Lee RG, Foerster J, Lukens J, et al., eds. Wintrobe’s clinical hematology, 10th ed. Philadelphia: Lippincott Williams & Wilkins, 1999:913, with permission.) |
demyelination.118, 119 The classic myelopathic syndrome is subacute combined degeneration, in which posterior and lateral column damage predominates; dorsal, pyramidal, and spinocerebellar tracts are affected. The earliest changes appear in the cervical or thoracic spine and can be detected by magnetic resonance imaging (MRI) as hyperintensity on T2-weighted images (Fig. 36.7). Larger, more heavily myelinated fibers tend to be affected most often.120
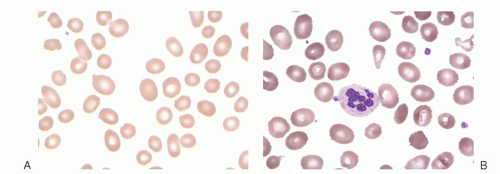 FIGURE 36.6. Blood smear from a patient with megaloblastic anemia due to cobalamin deficiency. Note characteristic large macro-ovalocytes (A) and hypersegmented neutrophils (B). (Courtesy of Irma Pereira MT [ASCP]SH.) |
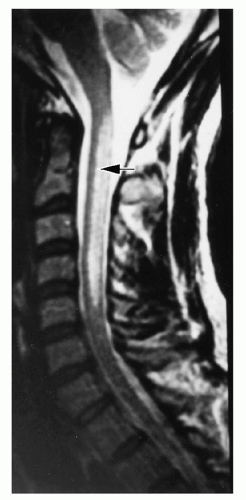 FIGURE 36.7. Magnetic resonance imaging (T2 weighted) of a sagittal section of the cervical spine of a man with pernicious anemia and severe myelopathy. Note the posterior localization of the high signal intensity lesion (arrow). (From Larner AJ, Zeman AZ, Allen CMC, et al. MRI appearances in subacute combined degeneration of the spinal cord due to vitamin B12 deficiency. J Neurol Neurosurg Psychiatry 1997;62:99-101, with permission.) |
only in retrospect after treatment. MRI reveals focal and diffuse changes in the brain138 (Fig. 36.8). Tensor diffusion imaging also shows white matter changes in adjacent areas that seem normal on MRI.139 Nevertheless, despite low cobalamin levels in 10% to 20% of patients with chronic dementias,95, 140 the low levels usually reflect only subclinical cobalamin deficiency (SCCD) and appear unrelated to chronic dementias, which rarely improve with cobalamin therapy.141 In very young children, however, some cobalamin deficiencies can cause developmental delay, lethargy, cerebral atrophy, and seizures.142
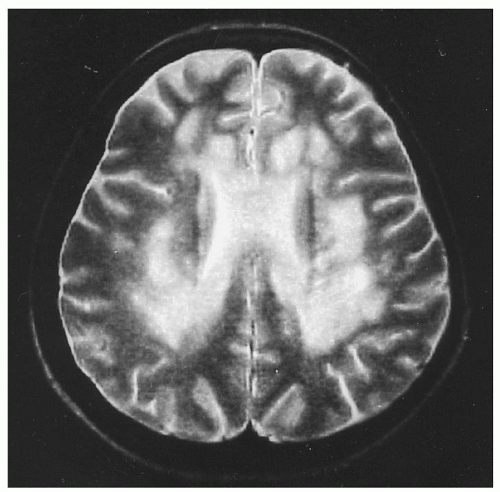 FIGURE 36.8. Magnetic resonance imaging (T2 weighted) of the brain in a woman with pernicious anemia and cognitive dysfunction. Large confluent and focal areas of increased signal intensities are seen, predominating around the ventricles. The changes improved after therapy. (From Stojsavljevic N, Levic Z, Drulovic J, et al. A 44-month clinical-brain MRI follow-up in a patient with B12 deficiency. Neurology 1997;49:878-881, with permission.) |
showed in 1985 that most of them reflected mild biochemical insufficiency that responded to cobalamin and, equally important, rarely involved IF-related malabsorption.94, 95, 96, 180 None of the subjects had cobalamin-related anemia although bone marrow cells displayed reversible metabolic defects. Similarly, none had clinical neurologic findings although some,96, 141, 181 but not all,182 displayed mild, reversible electrophysiologic changes.
TABLE 36.2 COMPARISON BETWEEN CLINICAL AND SUBCLINICAL COBALAMIN DEFICIENCY STATES | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
and 25 mg pyridoxine) slowed brain atrophy by 30%204 and reduced cognitive decline.205 Its important features were that the subjects were mildly cognitively impaired at baseline (which predicts likely progression, but avoids the irreversibility of dementia), the responders were hyperhomocysteinemic, and it is not clear which of the three vitamins was the beneficial one.
patients with myelofibrosis or chronic myelogenous leukemia. It is unclear whether iron deficiency can cause hypersegmentation.230 Neutrophil segmentation is normally greater in blacks than in whites.228
TABLE 36.3 CAUSES OF MACROCYTOSIS, DEFINED AS MEAN CORPUSCULAR VOLUME (MCV) GREATER THAN 97 FLa | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
or holo-TC II).190 Metabolic tests are essential in the diagnosis of inborn errors of metabolism, in which vitamin levels are often normal. Metabolites are also ideal for monitoring response of deficiency to therapy because their levels do not change unless the therapy was effective, whereas vitamin levels (serum cobalamin, holo-TC II, or folate) rise upon vitamin entry into the bloodstream regardless of efficacy. Metabolite improvement is also delayed for several days, allowing a post-therapy window of time for diagnostic reassessment if needed.
TABLE 36.4 BIOCHEMICAL TESTS FOR THE DIAGNOSIS AND DIFFERENTIATION OF CLINICALLY RELEVANT COBALAMIN AND FOLATE DEFICIENCIES | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


