FIGURE 7.1 Tumor progression through cooperation of proliferative and antiapoptotic functions. In normal cells in epithelial tissues (green cells) initiating mutational events such as deregulation of c-myc expression deregulate cell growth control and promote abnormal cell proliferation (yellow cells) while triggering a proapoptotic tumor suppression (red apoptotic cells) mechanism that can restrict tumor expansion. Subsequent acquisition of mutations that disables the apoptotic response, exemplified by Bcl-2 overexpression, prevents this effective means of culling emerging tumor cells, thereby favoring tumor expansion. Similar oncogenic events occur in lymphoid tissues.
The effectiveness of many existing anticancer drugs involves or is facilitated by triggering the apoptotic response. Thus, a detailed understanding of the components, molecular signaling events, and control points in the apoptotic pathway has enabled rational approaches to chemotherapy aimed at restoring the capacity for apoptosis to tumor cells. Identification of the molecular means by which tumors inactivate apoptosis has led to cancer therapies directly targeting the apoptotic pathway. These drugs are now being used in the clinic to specifically reactivate apoptosis in tumor cells in which it is disabled to achieve tumor regression.
Model Organisms Provide Mechanistic Insight into Apoptosis Regulation
Key to elevating the field of programmed cell death from a descriptive to a mechanism-based process was the discovery of genes in the nematode Ceanorhabditis elegans that control cell death, the cell death defective or ced genes.8 Genetic analysis revealed that ced-4 and ced-3 promote cell death, as worms with defective mutations in these genes possessed extra cells. In contrast, the ced-9 gene inhibited the death-promoting function of ced-4 and ced-3, thereby maintaining cell viability.9 ced-9 in turn was inhibited by egl-1, thereby promoting cell death. This creates a linear genetic pathway controlled upstream by cell-specific death specification regulators, and downstream by cell corpse engulfment and degradation mechanisms (Fig. 7.2).10 These findings helped propel work in mammalian systems when it became apparent that Ced-9 was homologous to BCL2,11 Ced-3 was homologous to interleukin1-β–converting enzyme, a cysteine protease that would later be classified as a member of the caspase family of aspartic acid proteases,12 Egl-1 was a BH3-only protein homologue,10 and that the proapoptotic factor apoptotic protease-activating factor (APAF-1)-1 identified in mammals was homologous to Ced-4.13
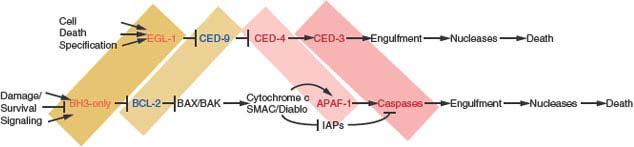
FIGURE 7.2 Analogous pathways regulate programmed cell death/apoptosis in metazoans. Regulation of programmed cell death in the nematode Ceanorhabditis elegans (top) and regulation of apoptosis in mammals (bottom). Shaded regions highlight corresponding homologous genes and protein families. In C. elegans, numerous cell death specification genes can up-regulate the transcription of the BH3-only protein Egl-1, which interacts with the antiapoptotic Bcl-2 homologue Ced-9 inhibiting is interaction with Ced-4. Ced-4, the Apaf-1 homologue, in turn, activates the caspase Ced-3, leading to cell death. A variety of engulfment gene products are then responsible for apoptotic corpse elimination and nucleases degrade the genome. In mammals, many survival, damage, and stress events impinge on the numerous members of the BH3-only class of proapoptotic proteins to either activate them to promote apoptosis or suppress their activation to enable cell survival. BH3-only proteins interact with and antagonize the numerous Bcl-2-related multidomain antiapoptotic proteins that serve to sequester proapoptotic Bax and Bak and may also contribute directly to Bax/Bak activation. Bax or Bak is essential for signaling apoptosis by permeabilizing the outer mitochondrial membrane to allow the release of cytochrome c and second mitochondrial-derived activator of caspase (SMAC). Cytochrome c acts as a cofactor for Apaf-1-mediated caspase activation in the apoptosome, and the SMAC amino-terminal four amino acids bind and antagonize the inhibitors of apoptosis proteins that interact with and suppress caspases, leading to their activation, widespread substrate cleavage, and cell death. Many engulfment gene products are responsible for corpse elimination and caspase-activated nucleases in the apoptotic cell itself, and additional nucleases within the engulfing cell are responsible for degradation of the genome.
A similar cell death pathway in the fruit fly Drosophila melanogaster identified Reaper, Hid and Grim as inhibitors of the inhibitors of apoptosis proteins (IAPs) that negatively regulate caspase activation. This eventually led to the identification of their mammalian counterpart second mitochondrial-derived activator of caspase (SMAC), also known as direct IAP-binding protein with low pI (DIABLO).14 These and other studies established the paradigm whereby proapoptotic BH3-only proteins inhibit antiapoptotic Bcl-2 proteins that prevent APAF-1-mediated caspase activation suppressed by IAPs, and the caspase-mediated proteolytic cellular destruction leads rapidly to cell death. It would later be realized that in mammals BH3-only proteins could also act as direct activators of the proapoptotic machinery (see later discussion).
Discovery of Bcl-2 and its Role as an Apoptosis Inhibitor in B-cell Lymphoma
To identify mechanisms of oncogenesis, the bcl-2 gene was cloned from the site of frequent chromosome translocation t(14;18):(q32;q21) in human follicular lymphoma.15–17 This chromosome rearrangement places bcl-2 under the transcriptional control of the immunoglobulin heavy chain locus causing abnormally high levels of bcl-2 expression. Distinct from other oncogenes at the time, instead of promoting cell proliferation, bcl-2 promoted B-cell tumorigenesis by the novel concept of providing a survival advantage to cells stimulated to proliferate by c-myc.18 Indeed, engineering high Bcl-2 expression in the lymphoid compartment in mutant mice promotes follicular hyperplasia that progresses to lymphoma upon c-myc translocation, and bcl-2 synergizes with c-myc to produce lymphoid tumors, paralleling events in human follicular lymphoma.19,20 Bcl-2 localizes to mitochondria where it has broad activity in promoting cell survival through suppression of apoptosis,21 provoked by numerous events, including oncogene activation (c-myc, E1A), tumor suppressor activation (p53), growth factor and cytokine limitation, and cellular damage. It also became clear that inactivation of the retinoblastoma tumor suppressor (Rb) pathway promotes a p53-mediated apoptotic response, suggesting that apoptosis was part of a tumor suppression mechanism that responded to deregulation of cell growth.6,22,23 Indeed, apoptotic defects acquired by a variety of means are a common event in human tumorigenesis.
Control of Apoptosis by Bcl-2 Family Members
Bcl-2 is the first member of what is now a large family of related proteins that regulate apoptosis and are conserved among metazoans including worms, flies, and mammals, and also viruses.24−28 Multidomain Bcl-2 family members containing Bcl-2 homology regions 1-4 (BH1-4) are either antiapoptotic (Bcl-2, Bcl-xL, Bcl-w, Mcl-1, and Bfl-1/A1, and virally encoded Bcl-2 homologues such as E1B 19K), or proapoptotic (Bax and Bak). Antiapoptotic proteins can block apoptosis by binding and sequestering Bax and Bak or by indirectly preventing Bax and Bak activation (Fig. 7.3).25,29,30
Bax and Bak are functionally redundant and required for signaling apoptosis through mitochondria, and deficiency in Bax and Bak produces a profound defect in apoptosis. Bax and Bak are considered the core apoptosis machinery controlled directly or indirectly by antiapoptotic Bcl-2-like proteins and proapoptotic BH3-only proteins. Remarkably, mice deficient in both Bax and Bak develop relatively normally, suggesting that other death mechanisms can compensate for loss of apoptosis in development.31 In healthy cells, Bak is bound and sequestered by Mcl-1 and Bcl-xL at cellular membranes, whereas Bax resides in the cytosol in a latent form and requires activation and translocation to membranes, where it is either sequestered by antiapoptotic Bcl-2-like proteins or otherwise induces apoptosis (Fig. 7.3).
Control of Multidomain Bcl-2 Family Proteins by the BH3-only Proteins
Bax and Bak deficiency abrogates the ability of BH3-only proteins to induce apoptosis, placing them upstream and dependent on the core apoptosis machinery.32 BH3-only protein Bcl-2 family members (Bim, Bid, Nbk/Bik, Puma, Bmf, Bad, and Noxa) are proapoptotic and antagonize the survival activity of antiapoptotic Bcl-2-like proteins by binding and displacing Bax and Bak to allow apoptosis (BH3-only proteins as neutralizers of Bcl-2) (Fig. 7.4).30 The different BH3-only proteins respond to specific stimuli to activate apoptosis (Fig. 7.3). For example, Bim induces apoptosis in response to taxanes, Puma and Noxa are transcriptional targets of and mediate apoptosis in response to p53 activation, Bad signals apoptosis on growth factor withdrawal, Nbk/Bik promotes apoptosis in response to inhibition of protein synthesis, and Bid is required for apoptosis signaled by death receptors. All of these signals are transduced from the BH3-only proteins to other members of the Bcl-2 family by protein-protein interactions.
The BH3 region of BH3-only proteins binds to a hydrophobic cleft in the multidomain Bcl-2-like antiapoptotic proteins that also supports Bax and Bak binding,33,34 causing their displacement (neutralization mode; Fig. 7.4).30 Differential binding specificities among the BH3 regions of the different BH3-only proteins determine whether they bind one or more Bcl-2-like proteins and displace Bax or Bak or both.35 Noxa binds and antagonizes Mcl-1, whereas Bad binds and antagonizes Bcl-2 and Bcl-xL, necessitating cooperation between Noxa and Bad function for efficient apoptosis. In contrast, Bim, Bid, and Puma have broader binding specificity and antagonize Mcl-1, Bcl-2, and Bcl-xL to release both Bax and Bak to induce apoptosis. Bim, the active form of Bid (truncated Bid or tBid) and possibly Puma can also be direct activators of Bax and Bak. For example, tBid can bind to latent, inactive Bax and promote its conformational change and translocation to the mitochondrial membrane that is required for apoptosis.36 BH3-only proteins can inhibit Bcl-2-like proteins, releasing these direct activators of Bax and Bak to promote apoptosis in the de-repression mode (Fig. 7.4). BH3-only proteins that only interact with Bcl-2-like proteins can release activator BH3-only proteins to promote apoptosis in the sensitizer mode (Fig. 7.4). Thus, apoptosis induction by BH3-only proteins can occur through neutralization, de-repression and sensitizer functions.28
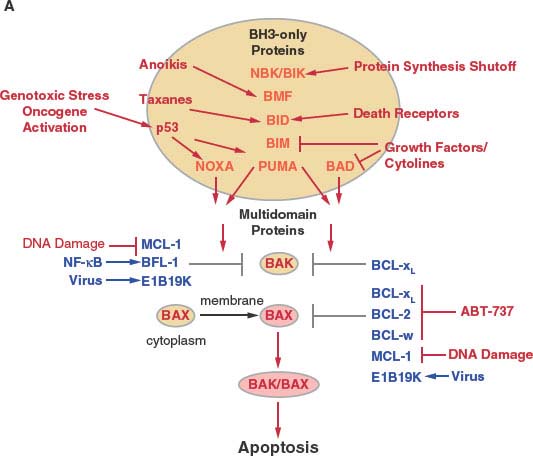
FIGURE 7.3 Regulation of apoptosis by the Bcl-2 family of proteins in mammals. A: Schematic of apoptosis regulation by the Bcl-2 family. Cytotoxic events activate, while survival signaling events suppress the activity of the BH3-only class of Bcl-2 family members (orange). BH3-only proteins are controlled at the transcription level and also by numerous posttranscriptional events that modulate phosphorylation, proteolysis, localization, sequestration, and protein stability. Once activated, BH3-only proteins disrupt functional sequestration of Bak and Bax by the multidomain antiapoptotic Bcl-2-like proteins (blue) and may also directly facilitate Bax/Bak activation. Although Bak is commonly membrane-associated in a complex with Mcl-1 and Bcl-xL in healthy cells, Bax resides in the cytoplasm as an inactive monomer with its carboxy-terminus occluding the BH3-binding hydrophobic cleft.138 Bax activation thereby additionally requires a change in protein conformation and membrane translocation by an unknown mechanism that may be facilitated by tBid binding. Binding specificity among BH3-only proteins for antiapoptotic Bcl-2-like proteins determines which complexes are disrupted, with some BH3-only proteins having broad specificity and others do not. Survival and death signaling events can also modulate apoptosis by targeting the multidomain antiapoptotic proteins either by antagonizing their function to promote apoptosis or induction their function to promote survival. ABT-737 is a rationally designed BAD BH3 mimetic that can bind Bcl-2, Bcl-xL, and Bcl-w but not Mcl-1 that can promote apoptosis where survival does not depend on Mcl-1. Once activated, Bax or Bak oligomerization promotes apoptosis. (continued)
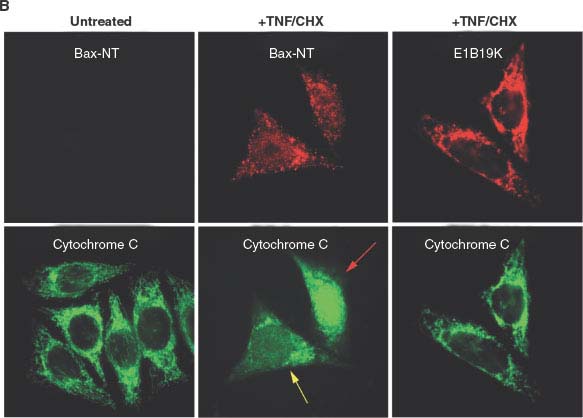
FIGURE 7.3 (Continued) B: Tumor necrosis factor-α (TNF-α) apoptotic signaling induces mitochondrial membrane translocation and a conformational change exposing the amino-terminus of Bax (visualized here by the Bax-NT antibody) and apoptosis, which is blocked by sequestration of Bax by the antiapoptotic viral Bcl-2 homologue E1B 19K. The human cancer cell line (HeLa cells) with or without E1B 19K expression, were then left untreated or treated with TNF/CHX. The localization of conformationally altered Bax (Bax-NT) and cytochrome c (left and middle panels), or E1B 19K and cytochrome c (right panel), are shown. The proapoptotic stimulus (TNF/CHX) induces Bax activation, mitochondrial translocation, and cytochrome c release from mitochondria that leads to caspase activation and apoptotic cell death, whereas expression of E1B 19K sequesters Bax thereby blocking cytochrome c release from mitochondria, caspase activation, and apoptotic cell death. The yellow and red arrows, respectively, mark cells with partial or complete cytochrome c release from mitochondria upon TNF/CHX treatment.
Importantly, it is this BH3 interaction with Bcl-2 that is the molecular basis for the BH3-mimetic class of proapoptotic, Bcl-2−antagonizing anticancer drugs (Fig. 7.5).33,37−39 This detailed understanding of the Bcl-2 family member protein interactions and function is allowing rational, apoptosis-targeted therapy (see later discussion).
Role of Mitochondrial Membrane Permeabilization in Apoptosis
Once activated, Bax and Bak oligomerize in the mitochondrial outer membrane, rendering it permeable to proapoptotic mitochondrial proteins cytochrome c and SMAC.40−44 How Bcl-2 family members permeabilize membranes is not entirely clear but it is likely related to a change in topology of the proteins in the membrane and formation of a channel or pore. Once released into the cytoplasm, cytochrome c interacts with the WD40 domains of APAF-1 in the apoptosome, a wheel-like particle with sevenfold symmetry that serves as a scaffold for caspase-9 activation.45 SMAC functions to antagonize the caspase inhibitors, the IAP proteins, to facilitate caspase activation. The amino-terminus of SMAC binds to IAPs, neutralizing their caspase-inhibitor function. Subsequent effector caspase activation (e.g., caspase-3), leads to the rapid, orderly dismantling of the cell and cell death without activating the innate immune response.46
Control of Apoptosis by Death Receptors
One of the apoptotic pathways being modulated in cancer therapies is that belonging to the death receptors. Ligands related to tumor necrosis factor-α (TNF-α), including Fas/Apo1 (Fas) and tumor necrosis factor-related ligand (TRAIL) and their cognate receptors were identified as potent activators of apoptosis, and this pathway is critical for regulating the immune response.47 Engagement of the receptor by soluble or membrane-localized ligand activates the death-inducing signaling complex composed of adaptor proteins such as FADD, which promotes activation of caspase-8 (Fig. 7.5). Caspase-8, in turn, cleaves the BH3-only protein Bid to its truncated or activated form tBid, which then antagonizes the antiapoptotic function of Bcl-2-like proteins, promoting Bax and Bak activation.36 This process signals cytochrome c and SMAC release from mitochondria and caspase-9 and -3 activation and cell death. In some cell types that do not require this Bcl-2 family protein-regulated, mitochondrial amplification step, active caspase-8 can directly cleave and activate caspase-3 to cause cell death by apoptosis. Execution of apoptotic cell death is extremely rapid and efficient, resulting in cell death in less than 1 hour in mammalian cells.
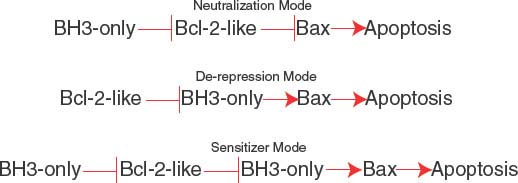
FIGURE 7.4 Modes of apoptosis activation by BH3-only proteins. The neutralization mode (top), de-repression mode (middle), and sensitizer mode (bottom). See text for explanation.
Modulation of the Death Receptor Pathway in Cancer Therapy
The ability of soluble ligands to activate the apoptotic response has stimulated interest in using this pathway to therapeutically induce apoptosis preferentially in tumor cells. Although TNF-α and Fas proved highly toxic to both normal and tumor cells, tumor cells display preferential sensitivity to TRAIL, which has now entered clinical trials (Fig. 7.6).48–52 Moreover, in cases in which apoptosis is blocked at the mitochondrial level in tumors, SMAC-mimetics have proved useful in stimulating the activity of TRAIL by antagonizing the caspase-inhibitory function of IAPs to facilitate direct caspase-3 activation by caspase-8 (Fig. 7.6).53–56 Thus, defining this pathway to apoptosis regulation has revealed novel opportunities to rational therapy designed to activate apoptosis preferentially in tumor cells.
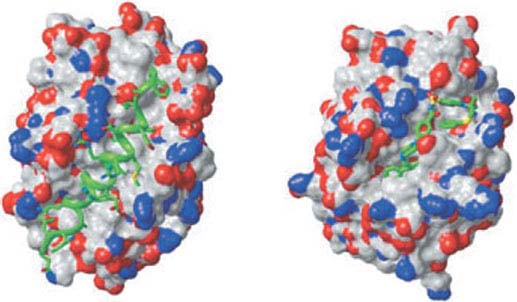
FIGURE 7.5 Three-dimensional structure of Bcl-xL with bound Bad BH3 ligand and ABT-737. Space-filling model of Bcl-xL illustrating the hydrophobic cleft binding the 25-mer peptide (green helix) of the Bad BH3 (left) or the rationally designed BH3-mimetic ABT-737 (right). (Reprinted from ref. 33, with permission.)
Drugs Targeting the Bcl-2 Family for Chemotherapy
In addition to Bcl-2 up-regulation in B-cell lymphoma as previously described, there are other mechanisms for directly or indirectly inactivating apoptosis in tumors that facilitate tumor progression and treatment resistance. Inactivation of the p53 tumor suppressor, or the p53 pathway through the gain of function of the p53 inhibitor MDM-2, is a common occurrence in tumors that results in the loss of the proapoptotic and growth arrest functions of p53.57,58 The BH3-only proteins Puma and Noxa are transcriptional targets of p53, the loss of which prevents induction of the p53-mediated response to genotoxic stress in tumors as a mechanism of tumor suppression. Various means for restoration of p53 function in tumors are, therefore, an attractive therapeutic approach.59–61
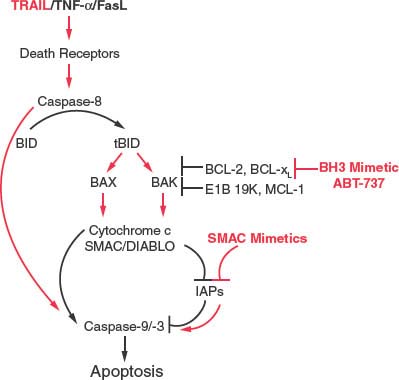
FIGURE 7.6 Therapeutic modulation of the apoptotic pathway downstream of death receptors. Tumor necrosis factor-related ligand (TRAIL) and related death-promoting ligands engage their cognate death receptors and activate caspase-8, which then cleaves Bid to active tBid. tBid can bind Bcl-2 and related antiapoptotic proteins to release Bax and Bak and may also directly promote their activation to permeabilize the mitochondrial outer membrane to release the APAF-1 cofactor cytochrome c, and the inhibitors of apoptosis protein antagonist second mitochondrial-derived activator of caspase (SMAC) for promote caspase-9 and -3 activation and cell death. BH3-mimetics such as ABT-737 can promote apoptosis-induction by TRAIL by relieving the protective capacity of the antiapoptotic Bcl-2-like proteins. In cells that do not depend on the mitochondrial apoptotic signal, TRAIL-mediated caspase-8 activation can directly promote downstream caspase activation and can synergize with SMAC mimetics in this case.
Activation of the MAP kinase pathway is also common in tumors and results in stimulation of tumor cell proliferation, but also the phosphorylation and proteasome mediated degradation of the BH3-only protein Bim. This Bim inactivation promotes tumor growth, while also producing resistance to the taxane class of chemotherapeutic drugs. This loss of Bim function is rectified by blocking Bim degradation with a proteasome inhibitor (bortezomib) (Fig. 7.7).62 Similarly, direct inhibition of MAP kinase pathway signaling with inhibitors (sorafenib, UO126) can also restore apoptotic function in addition to suppressing the proliferative response (Fig. 7.7). Receptor tyrosine kinase pathway activation in tumors also promotes tumor cell proliferation in part through MAP kinase pathway activation downstream and in part through Bim and thereby apoptosis inactivation. In chronic myelogenous leukemia in which chromosomal translocation and activation of the Bcr/Abl tyrosine kinase also leads to BIM inhibition, blocking kinase signaling with imatinib mesylate restores Bim and also Bad apoptotic function as a therapeutic strategy (Fig. 7.7).63 Activation of the PI-3 kinase pathway commonly through loss of PTEN tumor suppressor function and AKT activation results in phosphorylation and inactivation of the BH3-only protein Bad and reduction of Bim transcription through inhibition of forkhead factors, resulting in down-regulation of apoptosis.64 Thus, inhibitors of the PI-3 kinase pathway can restore apoptosis and facilitate tumor regression.65 NF-kB, a cytokine-responsive transcription factor, also promotes tumor growth while turning on the expression of antiapoptotic regulators Bcl-xL, Bfl-1 and IAPs (Fig. 7.3).66 Strategies to inhibit NF-kB are likely to promote tumor regression in part through restoration of apoptotic function.67
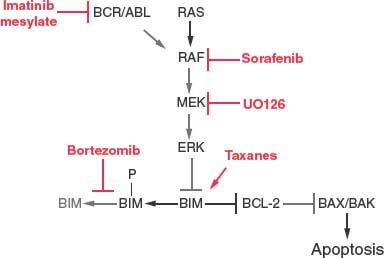
FIGURE 7.7 Therapeutic regulation of Bim and the MAP kinase pathway in cancer chemotherapy. Bim protein stability is regulated by Erk phosphorylation and proteasome-mediated degradation. Therapeutic modulation of the MAP kinase pathway (imatinib, sorafenib, and UO126) or proteasome function (bortezomib) can restore Bim protein levels and apoptosis function. Taxanes also stimulate Bim expression and promote Bim-mediated apoptosis, synergizing with the aforementioned inhibitors.
Direct Modulation of Bcl-2 with BH3-mimetics
The observation that antiapoptotic Bcl-2 family members bound and sequestered BH3-regions in a hydrophobic cleft as a means to suppress apoptosis activation (Fig. 7.5) revealed the opportunity for the rational design of small molecules that occlude the cleft and disrupt this Bcl-2-like protein/proapoptotic protein interaction, thereby promoting apoptosis.33 This was accomplished and resulted in ABT-737, which binds the BH3 binding pocket of Bcl-2, Bcl-xL, and Bcl-w, but not Mcl-1, similarly to the Bcl-2 family protein binding by Bad (Fig. 7.5). ABT-737 exhibited activity as a single agent against human lymphoma and small cell lung cancer cell lines in vitro and in mouse xenographs in vivo,37 and in combination with various cytotoxic agents against acute myeloid leukemia,68 multiple myeloma,69 chronic lymphocytic leukemia,70 and small cell lung cancer.71 ABT-263, an orally bioavailable form of ABT-737, exhibited similar preclinical activity,72 and has now entered clinical trials as a single agent or in combination with other anticancer drugs. Not surprisingly, given that ABT-737 does not bind to Mcl-1, resistance to ABT-737 has been associated with Mcl-1, as well as Bfl-1, up-regulation,73 indicating that combinatorial treatment with anticancer agents that target Mcl-174 or Bfl-1 could be therapeutically beneficial. Alternate chemical approaches to generating BH3-mimetics to promote apoptosis in cancer cells are also producing encouraging results in the preclinical setting.39 Thus, deciphering the mechanisms of apoptosis regulation in tumor cells is yielding novel opportunities for rational drug design and therapeutic intervention. These analyses can help predict which tumors have the potential to respond to apoptosis modulation and the types of drug combinations they may respond to.
Killing the Unkillable Cells: Alternate Approaches to Achieving Tumor Cell Death
An apoptotic response to therapy in tumors may not always be possible to achieve; therefore, it is important to determine alternate cell death processes and how to access them specifically in tumor cells. One intrinsic difference between normal and tumor cells is their altered metabolism and prevalence of aerobic glycolysis, which is an inefficient means for generation of adenosine triphosphate (ATP) required for sustaining homeostasis but an efficient means to generate synthetic precursors to support cell proliferation.75,76 This tumor cell-specific altered metabolism can provide novel approaches to therapeutically target cancer but not normal cells.75 Altered tumor cell metabolism is frequently coupled with high energy demand due to a rapid cell growth, with the potential to render tumor cells susceptible to cell death because of metabolic catastrophe where cellular energy consumption exceeds production.77
The means to specifically drive tumor cells toward metabolic catastrophe is through therapeutic nutrient deprivation that may be an additional consequence of the use of angiogenesis, growth factor, nutrient transporter, and metabolic pathway inhibitors. In addition, inhibition of the catabolic process of autophagy may similarly create metabolic deprivation and promote tumor cell death. Importantly, induction of cell death by interfering with metabolism can occur independently of an intact apoptotic response, suggesting that modulation of tumor cell metabolism may be therapeutically advantageous.
AUTOPHAGY
Role of Autophagy in Promoting Cell Survival to Metabolic Stress
Autophagy is an evolutionarily conserved, stress-activated catabolic lysosomal pathway that results in degradation of long-lived proteins and organelles. This process involves formation of the “autophagosome,” a double-membrane vesicle in the cytosol that engulfs organelles and cytoplasm and then fuses with the lysosome to form the “autolysosome,” where the sequestered contents are degraded and recycled to generate building blocks for macromolecular synthesis and maintenance of energy homeostasis.78,79 Hence the name autophagy (commonly pronounced aw-tof´ə-je), a term coined from the Greek auto or oneself, and phagy or eating, accurately depicts the process. Although autophagy can potentially induce cell death through progressive cellular consumption (autophagy is sometime referred to as type II programmed cell death), physiologic conditions in mammals where this occurs have not yet been identified. In most settings, autophagy is a survival pathway that can delay apoptosis, support metabolism in nutrient stress, and mitigate cellular damage by preventing the accumulation of damaged proteins and organelles. In tumor cells with defects in apoptosis, it has recently become apparent that autophagy supports long-term survival of tumor cells,80,81 newly revealing opportunities to target not only apoptosis, but also the mechanism by which cells survive once apoptosis is disabled.82
Autophagy is regulated by mTOR in the PI3-kinase/AKT pathway that functions to link nutrient availability to cellular metabolism.83 Under conditions of nutrient limitation, normal cells use this pathway to turn down growth and protein synthesis while activating the catabolic process of autophagy to maintain energy and biosynthetic homeostasis.79 Thus, autophagy is a temporary survival mechanism during starvation, as self-digestion provides an alternative energy source.80,84–86
On growth factor deprivation, hematopoietic cells activate autophagy, which is essential for maintenance of ATP production and cellular survival.81 In normal mouse development, amino acid production by autophagic degradation of “self” proteins allows maintenance of energy homeostasis and survival during neonatal starvation.87 Chronically ischemic myocardium induces autophagy, which inhibits apoptosis and may function as a cardioprotective mechanism.88 These and other examples indicate that autophagy is essential for the maintenance of cellular energy homeostasis that enables survival particularly during stress and starvation.
Autophagy is not only involved in recycling of normal cellular constituents to support cellular metabolism, but is also essential for the removal of toxic-damaged proteins and organelles. The importance of this toxic garbage disposal mechanism is exemplified by the observation that defects in autophagy result in the accumulation of ubiquitin-positive and p62-positive protein aggregates, abnormal mitochondria, and deformed cellular structures associated with production of reactive oxygen species and cellular degeneration.89–93 This protein and organelle quality control function of autophagy is important in preserving cellular health and viability in conjunction with metabolic support via catabolism.
Autophagy also contributes to innate immunity by protecting cells against infection with intracellular pathogens94–97 and to acquired immunity by promoting T-lymphocyte survival and proliferation98 and by affecting antigen presentation in dendritic cells and bacterial handling.99 Moreover, autophagy is involved in cellular development and differentiation,100,101 and may have a protective role against aging and age-related pathologies.102,103
Progressive autophagy can potentially lead to cell death in limited circumstances when allowed to proceed to completion, and when cells unable to undergo apoptosis are triggered to die. Unfortunately, it is often unclear whether autophagy is directly involved in initiation and/or execution of cell death or if it merely represents a failed or exhausted attempt to preserve cell viability.104 Recent studies indicate that autophagy may play an active role in programmed cell death, but the conditions under which autophagy promotes cell death versus cell survival remain to be resolved.105–108
Role of Autophagy in Tumorigenesis
Defective autophagy has been implicated in tumorigenesis, as the essential autophagy regulator becn1 is monoallelically deleted in human breast, ovarian, and prostate cancers109 and some human breast cancers have decreased Beclin1 levels.110 Becn1 is the mammalian orthologue of the yeast atg6/vps30 gene, which is required for autophagosome formation.111 Becn1 complements the autophagy defect present in atg6/vps30-disrupted yeast and in human MCF7 breast cancer cells, the latter in association with inhibition of MCF7-induced tumorigenesis in nude mice.110 Becn1−/− mice die early in embryogenesis, whereas aging Becn1+/− mice have increased incidence of lymphoma and carcinomas of the lung and liver112,113 In addition, mammary tissue from Becn1+/− mice shows hyperproliferative, preneoplastic changes.113 Tumors forming in Becn1+/− mice express wild type Beclin1 mRNA and protein, indicating that Becn1 is a haploinsufficient tumor suppressor.112
Recent studies revealed that autophagy enables tumor cell survival in vitro and in vivo that is particularly obvious in tumor cells when apoptosis is inactivated.80,85,86 When angiogenesis is insufficient, autophagy localizes to the resulting hypoxic tumor regions where it supports tumor cell viability (Fig. 7.8).80,85,86 This process of autophagy during nutrient deprivation allows recovery of growth and proliferative capacity with remarkably high fidelity when nutrients, oxygen, and growth factors are restored. Thus, autophagy may be a fundamental obstacle to tumor eradication.80
How inactivation of a survival pathway promotes tumorigenesis has been an intriguing question. The apparently conflicting pro-survival and pro-death functions of autophagy can, however, be reconciled if one considers autophagy a prolonged but interruptible pathway to cell death on stress and starvation, where nutrient restoration prior to its culmination can provide cellular salvation. This contrasts the death processes of apoptosis and necrosis, which are executed rapidly and are irreversible. As such, the identification of the precise mechanism by which autophagy supports survival is critical.
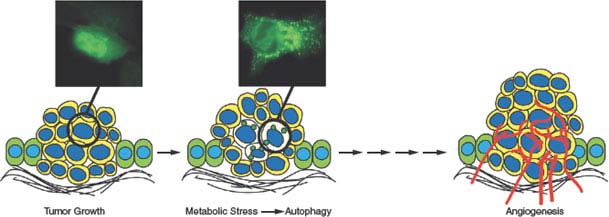
FIGURE 7.8 Role of autophagy in enabling survival of tumor cells to metabolic stress. As epithelial tumor cells proliferate and multiple cell layers accumulate, the initial absence of a blood supply produces metabolic stress in regions most distal to the supply of nutrients and oxygen often in the center of the tumor mass. In tumor cells with apoptosis defects, this allows tumor cells in these metabolically stressed tumor regions to survive through autophagy. Subsequent angiogenesis relieves metabolic stress, obviating the need for autophagy, fueling tumor growth.
Autophagy not only provides an alternate means for energy generation during periods of starvation, but also has a role in cellular damage mitigation through promotion of protein and organelle quality control, especially under conditions of stress in which proteins and organelles become damaged. By degrading damaged proteins and organelles, autophagy prevents their accumulation, which can be toxic. This function of autophagy is particularly critical in stressed tissues and tumors, which are regularly subjected to metabolic stress by their dependence on the inefficient process of aerobic glycolysis and by their intermittently limited blood supply during rapid tumor growth or metastasis. Thus, autophagy defects in tissues and tumors reduce cellular fitness and render cells prone to DNA damage, mutation, and genomic instability,85,86,92 which in turn contribute to tumor initiation and progression.82,114,115
In addition to these cell-autonomous mechanisms, defective autophagy can promote tumorigenesis in a non–cell-autonomous way by reducing tumor cell survival that causes chronic cell death and inflammation in tumors.80,92,115 Autophagy can thereby be thought of as a double-edged sword. On the one hand, autophagy promotes tumor cell survival through maintaining energy homeostasis and mitigating oxidative damage by preventing the accumulation of aggregated proteins and abnormal organelles. On the other hand, defects in autophagy elevate oxidative stress, DNA damage, and mutation, and promote chronic cell death and inflammation, all of which are linked to promotion of cancer initiation and progression.115 These observations have led to the notions that autophagy stimulation can prevent cancer, whereas inhibiting autophagy-mediated survival is an approach to treating established, aggressive cancers.82,114
Autophagy Modulation for Cancer Treatment
As autophagy can enable tumor cell survival to stress,80,81 the means to block autophagy has the potential to promote cell death that may be therapeutically advantageous.82,114 Moreover, both targeted and cytotoxic antineoplastic agents have been observed to induce autophagy in human cancer cell lines,116,117 possibly as a survival mechanism in response to treatment-induced stress. Thus, autophagy inhibitors are expected to deprive cancer cells of an essential survival mechanism and consequently render them more susceptible to cell death. This novel paradigm in cancer therapy has been validated in several preclinical studies.118–126 This approach is now under investigation in phase 1/2 clinical trials involving autophagy inhibition by the antimalarial drug hydroxychloroquine, which blocks lysosome degradation of the products of autophagy, in combination with standard chemotherapy.
NECROSIS
Recent evidence suggests that tumor cells in which apoptosis has been disabled can be diverted to necrosis, which has traditionally been considered an unregulated (and thus, not programmed) form of cell death implicated in pathologic states, such as ischemia, trauma, and infection.127 Recent evidence is calling the unregulated nature of necrosis into question.
Necrosis is derived from the Greek word nekros for corpse, and it involves rapid swelling of the cell, loss of plasma membrane integrity, and release of the cellular contents into the extracellular environment, resulting in an acute inflammatory response. Necrosis is largely viewed as an accidental and unregulated cellular event triggered by cellular trauma (direct physical injury), acute energy depletion, or extreme stress.128 Recently, a type of programmed necrotic cell death, called necroptosis,129 has been identified as induced by interaction of death domain receptors with their respective ligands under conditions of defective or inhibited downstream apoptotic machinery.130 Necroptosis depends on the serine/threonine kinase activity of the death domain receptor-associated adaptor Rip1 and its relative Rip3.131–136 Necroptosis is potently inhibited by the Rip1 kinase inhibitors, the necrostatins.133 In a genome-wide siRNA screen for necroptosis regulators, a set of 432 genes with enriched expression in the immune and nervous systems was identified and cellular sensitivity to necroptosis was found to depend on the same signaling network that mediates innate immunity.131 Harnessing necroptosis to induce cell death in cancer is an exciting new prospect, the exploitation of which will require a deeper mechanistic insight into the process and its regulation.
Regarding necrosis as a therapeutic end point, tumor cell fate in response to treatment with DNA-damaging agents depends on the effect of the DNA repair protein poly(ADP-ribose) polymerase (PARP) on cellular metabolism. PARP activation by DNA-damaging alkylating agents causes PARP-mediated β-nicotinamide adenine dinucleotide (NAD) consumption, ATP depletion, and metabolic stress. The glycolytic state (Warburg effect) and inefficient mode of energy production in most cancer cells renders them sensitive to this ATP depletion in response to PARP activation, resulting in induction of necrotic cell death of apoptosis-defective tumor cells.137 Tumor cells with defects in both apoptosis and autophagy may be particularly susceptible to death by necrosis as loss of autophagy potential deprives cells of an alternate energy source for maintenance of metabolism and viability in metabolic stress that is compounded by the Warburg effect.80
Manipulation of tumor cell metabolism is an appealing therapeutic approach, as it can be used to induce cancer cell death by metabolic catastrophe.77 This is particularly relevant for tumors with increased proliferative capacity and high bioenergetic requirements, such as tumors with constitutive activation of the PI3-kinase/Akt pathway, which are unable to down-regulate metabolism and to activate autophagy in response to starvation. Thus, the very properties that confer cancer cells with the capacity for rapid growth may also render them susceptible to metabolic stress pharmacologically induced by a wide variety of means, including nutrient deprivation, angiogenesis inhibition, glycolysis inhibition, accelerated ATP consumption, or autophagy inhibition. Furthermore, necrotic cell death can be genetically determined indirectly through manipulation of cellular bioenergetics (decreased energy production through autophagy and catabolism inhibition, increased metabolic demand through elevated consumption, or decreased nutrient availability) or directly by activating Rip kinases or PARP. It will be of great interest to see if necrosis, like apoptosis, can be exploited for cancer therapy.
It is becoming clear that cells possess multiple death mechanism, and establishing how these are altered in tumors and can be activated with therapy is essential. Defining at the molecular level how apoptosis is regulated has led to the development of novel cancer therapies aimed at triggering or restoring apoptotic function in tumor cells, and this progress is likely to continue. Moreover, defining the mechanisms by which common mutations in human tumors inactivate apoptosis has yielded novel opportunities for tumor-genotype–specific rational chemotherapy targeting the apoptotic pathway. In tumor cells where apoptosis is disabled, it is apparent that alternate forms of cell death can be activated, including necrosis, the process by which remains poorly characterized. Finally, the catabolic process of autophagy can promote tumor cell survival to metabolic stress, providing new opportunities for therapeutic intervention, in part capitalizing on the altered metabolic state intrinsic to tumor cells.
Selected References
The full list of references for this chapter appears in the online version.
3. Rao L, Debbas M, Sabbatini P, Hockenbery D, Korsmeyer S, White E. The adenovirus E1A proteins induce apoptosis, which is inhibited by the E1B 19-kDa and Bcl-2 proteins. Proc Natl Acad Sci U S A 1992;89:7742.
4. Fanidi A, Harrington EA, Evan GI. Cooperative interaction between c-myc and bcl-2 proto-oncogenes. Nature 1992;359:554.
5. Evan GI, Wyllie AH, Gilbert CS, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992;69:119.
6. Morgenbesser SD, Williams BO, Jacks T, DePinho RA. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature 1994;371:72.
7. Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006;9:351.
9. Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature 1992;356:494.
12. Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 1993;75:641.
13. Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997;90:405.
15. Bakhshi A, Jensen JP, Goldman P, et al. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell 1985;41:899.
16. Cleary ML, Smith SD, Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell 1986;47:19.
17. Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science 1985;229:1390.
18. Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988;335:440.
19. McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature 1991;349:254.
20. Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 1990;348:331.
22. Debbas M, White E. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev 1993;7:546.
23. Lowe SW, Ruley HE. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev 1993;7: 535.
31. Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001;292:727.
32. Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev 2001;15:1481.
34. Muchmore SW, Sattler M, Liang H, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1996;381:335.
37. Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005;435:677.
40. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000;102:33.
42. Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997;275:1132.
43. Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000;102:43.
44. Yang J, Liu X, Bhalla K, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 1997;275:1129.
56. Fulda S, Wick W, Weller M, Debatin KM. Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med 2002;8:808.
59. Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007;445:656.
60. Ventura A, Kirsch DG, McLaughlin ME, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007;445:661.
62. Tan TT, Degenhardt K, Nelson DA, et al. Key roles of BIM-driven apoptosis in epithelial tumors and rational chemotherapy. Cancer Cell 2005;7:227.
68. Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006; 10:375.
80. Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006;10:51.
81. Lum JJ, Bauer DE, Kong M, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005;120:237.
86. Mathew R, Kongara S, Beaudoin B, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007;21:1367.
87. Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032.
90. Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006;441: 880.
91. Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006;441:885.
92. Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062.
93. Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007;131:1149.
102. Lee JH, Budanov AV, Park EJ, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010;327:1223.
112. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003;100:15077.
113. Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003;112:1809.
114. White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 2009; 15:5308.
122. Degtyarev M, De Maziere A, Orr C, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol 2008;183:101.
123. Maclean KH, Dorsey FC, Cleveland JL, Kastan MB. Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest 2008;118:79.
124. Carew JS, Nawrocki ST, Kahue CN, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007;110:313.
125. Amaravadi RK, Yu D, Lum JJ, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007;117:326.
130. Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005;1:112.
133. Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008;4:313.
135. Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009; 137:1112.
136. He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009;137:1100.
138. Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 2000;103:645.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


