MEASLES AND RUBELLA
DIANE E. GRIFFIN
Measles and rubella are rash diseases of childhood that can be complicated by neurologic disease. Measles virus (MeV)–induced neurologic disease is associated with community-acquired infection of children, whereas the most important rubella virus (RV)–induced neurologic disease is associated with congenital infection. For both diseases, effective and safe vaccines exist. Despite wide distribution of measles vaccine, measles remains a frequent cause of morbidity and mortality worldwide, and even a low incidence of measles-associated neurologic disease has a significant impact on society because of the long-term neurologic disabilities that ensue (1). Rubella vaccine is less widely distributed, but there is an intensifying effort to increase coverage in developing countries and reduce the worldwide incidence of congenital rubella syndrome (CRS) (2). The World Health Organization has targeted both viruses for elimination and potential eradication (3).
Neurologic complications may result from direct virus invasion of the central nervous system (CNS) or from induction of an autoimmune response to CNS antigens. For measles, there are three distinct neurologic diseases that occur either at the time of the acute disease or months to many years after apparent recovery: acute disseminated encephalomyelitis (ADEM), measles inclusion body encephalitis (MIBE), and subacute sclerosing panencephalitis (SSPE). Rubella causes ADEM and progressive rubella panencephalitis (PRP) less frequently than measles but is also teratogenic and causes CRS.
BACKGROUND ON MEASLES
MeV, the etiologic agent of measles, is a member of the Morbillivirus genus of the Paramyxoviridae family of nonsegmented, negative-stranded, enveloped RNA viruses. There are several morbilliviruses and each has a relatively restricted host range. Nonprimate morbilliviruses cause respiratory disease in dogs, horses, cows, goats, sheep, and marine mammals and neurologic complications are common. Rinderpest, a disease of cattle, has recently been eradicated (4,5).
Morbilliviruses have six structural proteins (Fig. 8.1). The hemagglutinin (H) and fusion (F) are transmembrane proteins present on the surface of the virus and infected cell. These proteins are important for viral attachment and penetration of the target cell. The matrix (M) protein is found on the inner surface of the membrane and interacts with the cytoplasmic tails of H and F and with the nucleocapsid for virion assembly and budding. The nucleocapsid (N) protein surrounds and encapsidates the viral RNA to form the helical nucleocapsid structures. The phosphoprotein (P) and large (L) polymerase protein are also associated with the nucleocapsid and complete the viral elements necessary for RNA transcription. Two nonstructural proteins, C and V, are encoded within the P gene and regulate the host innate response to infection (6–12).
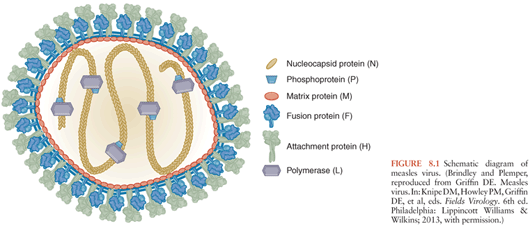
MeV transmission from person to person is by the respiratory route. The virus spreads from the initial site of replication in the respiratory tract to local draining lymph nodes (13–15). Replication in lymphatic tissue produces virus that then spreads through the blood to multiple organs including skin, lung, liver, spleen, and lymph nodes. The viremia is cell associated with infected B cells, T cells, and monocytes in circulation (16–18). MeV-infected leukocytes increase expression and activation of the integrins lymphocyte function-associated antigen (LFA)-1 and very late antigen (VLA)-4 that promote adherence to the surface of endothelial cells and this interaction is likely to facilitate the spread of infection to many organs and tissues (19,20). In tissue, endothelial and epithelial cells are also targets for infection (21–23).
Three cellular receptors have been identified: membrane cofactor protein or CD46 (24,25), signaling lymphocyte activation molecule (SLAM) or CD150 (26), and nectin 4 (27,28). CD46 is a widely distributed human complement regulatory protein expressed on all nucleated cells including the apical surface of polarized epithelial cells. In the CNS, CD46 is expressed on choroid plexus epithelial cells, on cerebral endothelium, and ependymal cells (29,30). SLAM/CD150 is a membrane glycoprotein expressed on cells of the immune system including immature thymocytes, activated T and B lymphocytes, activated monocytes, and mature dendritic cells (31,32) but is not expressed by brain parenchymal cells (29). Nectin 4 is an adherens junction protein expressed by epithelial cells in the lungs, tonsils, and placenta (33,34). Studies of different strains of MeV have shown that both vaccine and wild type viruses can use CD150 and nectin 4 as a receptor. However, although most vaccine strains use CD46 efficiently, wild type strains do not (35–37). The receptors used for neural cell infection by wild type MeV and chick cell infection by vaccine virus (38–40) have not been identified.
Through expression of the H and F proteins on the cell surface, infected cells may fuse with nearby cells to form syncytia or giant cells both in vitro and in the lungs and lymphoid tissues of infected patients. However, MeV is not detectable by usual pathologic or immunocytochemical techniques in the brains of patients dying acutely with measles (21). Studies using in situ hybridization have identified MeV infecting cerebral capillary endothelial cells during acute fatal disease (22). In addition, electroencephalographic (EEG) abnormalities and a cerebrospinal fluid (CSF) pleocytosis are common in acute uncomplicated measles (41–43), suggesting the possibility that MeV infection of the CNS is common during uncomplicated infection. However, these changes are also observed after measles immunization, so it is not clear that they indicate virus infection of the CNS (44).
The characteristic morbilliform rash of measles is due to immune cell infiltration into sites of MeV infection of skin epithelial cells and marks the onset of the immune response and the initiation of virus clearance (45). It is a time of intense immune activation (46–48) which is accompanied by suppression of skin test responses to recall antigens such as tuberculin (49,50) and decreased in vitro lymphoproliferative responses to mitogens (51). This immune suppression contributes to an increased susceptibility to secondary infections, the most common cause of death due to measles (52). The immune activation accompanying measles may contribute to the neurologic complications as well.
Although clearance of infectious MeV is generally complete after the rash has resolved, clearance of viral RNA requires many months (53,54). This continued presence of MeV RNA in lymphoid tissue and circulating mononuclear cells may contribute to immune suppression and to development of acute and chronic neurologic disease.
OVERVIEW OF THE NEUROLOGIC COMPLICATIONS OF MEASLES
The neurologic complications of measles are uncommon and occur at three distinct times in relation to the primary infection and acute disease (Fig. 8.2). ADEM usually presents within 1 to 2 weeks of the appearance of the rash (55,56), MIBE within a few months (57,58), and SSPE several years after initial infection (59). The age and general immune status of the individuals susceptible to these complications are also distinct (Table 8.1). ADEM occurs in individuals who have apparently normal immune systems and are older than 2 years at the time of primary infection (52). The incidence is 1:1,000 cases of measles (52,60). MIBE occurs in immunosuppressed patients of any age (61–65). The incidence is approximately 1:10 cases of measles in immunocompromised children (66,67). SSPE occurs in immunologically normal individuals who have often had measles at younger than 2 years (59,68,69). The incidence is approximately 1:10,000 cases of measles (70).
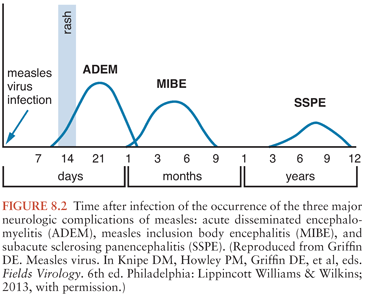
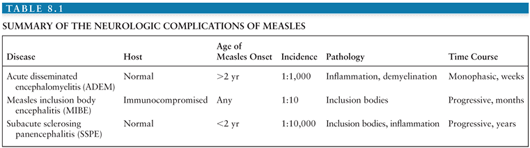
MeV is easily demonstrable in the brains of patients with MIBE and SSPE, but not ADEM (21). The current understanding of these three neurologic complications of measles is that ADEM is an autoimmune demyelinating disease triggered by measles, MIBE is a progressive MeV infection of the brain in patients unable to mount an effective immune response, and SSPE is a slowly progressive MeV infection of the CNS that is poorly controlled by the immune system and presents clinically many years after infection.
MEASLES INCLUSION-BODY ENCEPHALITIS
History
Progressive MeV infection was reported first by Hecht (71) in 1910 as a case of giant cell pneumonia. “Hecht pneumonia” was suspected to be a complication of measles and this hypothesis was proven in 1959 when Enders et al. (72) isolated MeV from the lungs and respiratory tract secretions of three young children with compromised immune systems and progressive pulmonary disease. Pathologic examination of the lungs of these children showed giant cell pneumonia. None had a history of rash or other classic signs or symptoms of measles, but all had histories of exposure to measles in the preceding months. Giant cells were found in other organs (e.g., liver, lymph nodes), but neurologic symptoms were not reported and it is not clear that the brain was examined (72).
Breitfeld and colleagues (73) first reported progressive nervous system disease in 1973 as cases of SSPE. Two young children with leukemia developed progressive neurologic disease and died approximately 6 months after exposure to measles. At autopsy, giant cells were not present in the CNS, but there were intranuclear and intracytoplasmic inclusions now recognized to be frequent pathologic features of persistent MeV infection of the CNS. One also had Hecht pneumonia. Subsequently, this neurologic disease, MIBE, was distinguished from SSPE by its time of onset in relationship to measles, lack of inflammatory response in the brain (74), and occurrence in immunocompromised individuals. Recently, this disease has also been described in adults who are immunocompromised due to HIV infection (65) and after immunization with the live attenuated MeV vaccine (75).
Pathogenesis and Pathology
The rash marks the onset of the immune response with the appearance of MeV-specific antibody and cellular immunity, and this immune response to MeV is normally effective at clearing infectious virus from blood and tissues (53). In individuals with severe acquired or genetic defects in cellular immunity, a rash may not appear (72,73) and MeV infection may not be controlled. These patients often develop progressive pulmonary or neurologic disease due to unrestrained MeV replication (66,73,74).
Virus infection of the CNS is presumed to begin with infection of cerebral capillary endothelial cells or with infiltration of infected leukocytes into the brain (20,22,76). Neurons or glial cells subsequently become infected and virus spreads slowly within the CNS. Intranuclear and intracytoplasmic eosinophilic inclusion bodies are seen primarily in gray matter areas (73). There is focal necrosis, and neurons often show signs of degeneration (74). The areas of brain most frequently involved are the parietooccipital areas, basal ganglia, and brainstem. Typically, there is little evidence of an inflammatory response to the infection, although proliferation of astrocytes and microglia is often apparent (61,74).
On electron microscopic examination, inclusion bodies contain the microtubular structures of MeV nucleocapsids (58,74). MeV antigens (particularly N) can be identified by immunocytochemical staining (77). However, the H, F, and M proteins are undetectable (78) and often MeV cannot be recovered in culture from brain tissue (61,64). Examination of viral RNA extracted from the brains of MIBE patients has shown that messenger RNAs (mRNAs) for the envelope proteins are limited in amount and that the virus has accumulated many mutations, particularly in the M-protein gene, similar to those found in SSPE (see later discussion) (79,80). These mutations often preclude or severely limit productive replication (81–83). These mutations may reflect positive selection and adaption of the virus to growth in neurons or frequent transcription errors, including biased hypermutation due to the action of dsRNA–dependent adenosine deaminase (84), combined with a lack of selection for replication-competent virus. Production of infectious virus may not be important for virus replication and spread in the CNS, because neurons can allow transsynaptic spread of viral RNA–containing nucleocapsids from cell to cell without production of infectious virions (85–88).
Clinical Manifestations
MIBE usually presents 1 to 6 months after exposure to measles with progressive neurologic deterioration in the absence of fever. Signs and symptoms include altered mental status, lethargy, slurred speech, focal motor seizures or epilepsia partialis continua, weakness, and occasionally blindness or hearing loss (57,62,65,77). Typically, the seizures are resistant to control by anticonvulsant medications (62). Disease progresses over days to weeks to coma and death and may be accompanied by inappropriate secretion of antidiuretic hormone (58,66,73).
Diagnosis
The diagnosis is often difficult because there may be no history of a rash. History of exposure to measles or immunization in the preceding months should be sought in immunosuppressed individuals with progressive neurologic deterioration. Brain biopsy with pathologic examination and reverse transcriptase-polymerase chain reaction (RT-PCR) to detect MeV RNA is often necessary for definitive diagnosis (62,65,89). At the time of presentation, there is often no detectable antibody to MeV, consistent with the poor immune response to infection, although at later times antibody may be present in serum or CSF (62,66,74). CSF examination is usually within normal limits, but occasionally there is a moderate elevation in the protein concentration (62). EEG findings are usually abnormal, but nonspecific and nondiagnostic, with diffuse slowing or periodic lateralized spike and wave activity (62,65,66,90). Computed tomographic (CT) scan and magnetic resonance imaging (MRI) scans are often normal at the time of presentation with later development of T2 signal abnormalities, edema, cortical atrophy, and ventricular dilation (62,65,66).
Treatment and Prevention
Adequate measles immunization prior to the onset of leukemia or other immunosuppressive illness undoubtedly prevents many cases of MIBE (57). It is difficult to discern the usefulness of postexposure immunoglobulin prophylaxis for immunocompromised individuals. Several cases of MIBE have occurred despite administration of immunoglobulin, but many more may have been prevented. There is no established antiviral treatment for MIBE and essentially all cases are fatal. However, there are case reports of neurologic improvement with prolonged intravenous ribavirin treatment (62) and slowed disease progression with initiation of antiretroviral therapy in patients with AIDS (65).
SUBACUTE SCLEROSING PANENCEPHALITIS
History
SSPE was first described in 1933 by Dawson (91) in a 16-year-old boy with progressive neurologic deterioration characterized by failing memory, slow and deliberate movements, and myoclonus. The following year, he reported an additional case of this subacute inclusion body encephalitis in a 5-year-old girl (92). Histologic examination of the brains of these patients showed inflammation with eosinophilic intranuclear and intracytoplasmic inclusions in neurons. The disease became known as Dawson encephalitis, and Dawson postulated that the disease was of viral etiology, but he could not transmit the disease to experimental animals. In 1945, van Bogaert (93) described a similar condition, subacute sclerosing leukoencephalitis with prominent white matter involvement. It was appreciated subsequently that the same disease could involve both gray and white matter (94). In 1966, paramyxovirus-like particles suggestive of MeV were seen on electron microscopic examination of the inclusions (95). Reports of these virus particles were followed rapidly by observations of elevated MeV antibody in serum and CSF, staining of the inclusions with antibody to MeV antigens and culture of MeV from brains (96–100).
Epidemiology
SSPE is a rare (approximately 1 in 10,000) late complication of measles (68,70,101). The mean time to onset of SSPE after measles is 6 to 10 years (59,102,103) (Fig. 8.2). Children with SSPE often have a history of acquiring measles at an early age (59,68–70,104–107) when the immune system is immature and maternal antibody may still be present. In most parts of the world, the disease occurs preferentially in boys (68,104,108,109). Exposure to birds has been identified as a risk factor (69,105,108). How these factors increase the risk of developing SSPE is unknown. There is no clustering of cases to suggest that the virus causing the initial infection leading to SSPE is different from the virus causing uncomplicated disease.
Pathogenesis and Pathology
SSPE is the most extensively studied of the neurologic complications of measles. Nevertheless, the pathogenesis of this rare complication remains obscure. It is not known whether infection of the nervous system occurs at the time of primary infection and progresses slowly with clinical evidence of disease apparent only after years or whether the infection is latent at a site outside the CNS and then spreads to the brain. The route of virus entry into the CNS is unknown, but infection of cerebral capillary endothelial cells is a likely possibility (76,11). Extensive sequence analysis of viral RNA from various parts of the brain in SSPE suggests that the virus in the nervous system is clonal (111), implying that virus entered the brain at one time and then gradually spread throughout the nervous system. This gradual spread has also been suggested by serial MRI studies (112,113). Therefore, it is most likely that MeV enters the brain at the time of the original acute infection or during the prolonged phase of circulating viral RNA and subsequently spreads slowly through the CNS eventually infecting a sufficient number of cells to produce dysfunction and clinical evidence of infection.
At the time that neurologic symptoms are recognized, the infection is extensive. Neurons and oligodendrocytes contain nuclear and cytoplasmic viral inclusion bodies (114), antibody responses are vigorous and evident both in serum and CSF (115,116), and there is an extensive mononuclear inflammatory reaction in the brain (92,93,117). Gray matter is most prominently affected, but pathologic changes are present in white matter as well (92,93). Retinitis is frequently present with MeV antigen demonstrable in the retinal neuroepithelium. At autopsy, MeV RNA or antigen can be detected in a wide variety of tissues (118).
Pathologic examination of the brain shows intranuclear and intracytoplasmic inclusions, in situ hybridization shows MeV RNA, and immunocytochemical staining shows MeV antigens (110). However, no virus is seen budding from the surface of infected cells (119). Nuclear inclusions are filled with “smooth” nucleocapsids (114,119) consistent with the absence of the associated L and P proteins necessary for transcription and replication of viral RNA. The cytoplasm contains replication-competent “fuzzy” nucleocapsids that extend into neuronal processes further suggesting that virus can spread within the CNS by cell-to-cell synaptic transmission of the ribonucleoprotein complex (85,87,88,113,120). The observation that strains of MeV isolated from SSPE patients are more likely than standard strains of MeV to cause neurologic disease after intracerebral inoculation into small animals and primates suggests that the virus has adapted to growth in neural tissue.
Like MIBE, the virus present in the brain of SSPE patients is replication defective and cannot usually be recovered from SSPE brain in a cell-free form (99,100,121). Extensive sequence analysis of viral RNAs has shown that SSPE viruses are of the same lineage as viruses that cause acute measles but distinguishable in the genes encoding the M, F, and H proteins (112,122–129). Studies of brain-associated viral proteins and RNA in SSPE have revealed differences in the relative amounts of the various viral mRNAs and proteins (127–130), in the antigenicity of viral proteins (131), and in RNA sequences coding for viral proteins (125–127) between SSPE viruses and wild type or vaccine strains of MeV. Because the disease cannot be studied prospectively, it is not known for certain whether these differences represent unique features of the original infecting virus, selection for growth in the CNS, or selection for growth in the presence of a vigorous antibody response.
In general, expression of M protein is low (130,132,133) and the mRNA encoding M extracted from SSPE brain is mutated throughout the gene. Construction of recombinant viruses has shown that functional M is dispensable for virus growth and spread in the CNS and may foster the formation of nuclear and cytoplasmic inclusion bodies (83). Mutations in the H and F envelope proteins that interfere with assembly and budding of infectious virus are also associated with persistent infection and SSPE (123,126,127,134).
The possibility that the development of SSPE represents a defect in the immune response has led to investigations of cellular and humoral immune responses to MeV and other antigens. In contrast to patients with MIBE, there is often an intense perivascular mononuclear inflammatory response in brain and high levels of antibody to MeV in serum. There is also significant production of MeV-specific antibody by plasma cells residing in the CNS. This locally synthesized antibody appears in the CSF leading to characteristic elevations in the level of CSF immunoglobulin much of which is MeV specific (96,135–137). Antibody produced in the CNS derives from clones of resident antibody-secreting B cells and is therefore of restricted heterogeneity leading to the appearance of oligoclonal immunoglobulin bands on electrophoretic analysis of the CSF from SSPE patients (138–142). Antibodies against the N and P proteins are particularly abundant and antibody against the M protein is particularly deficient (132,143). Antibody to CD9, a tetraspanin protein widely expressed in the CNS, is also elevated, raising the possibility of an autoimmune component to this progressive disease (144).
Experiments in small mammals have shown that treatment with antibody after intracerebral infection with neuroadapted strains of MeV attenuates acute disease but increases the incidence of persistent virus infection and subacute or chronic encephalitis (81,145). Cases of SSPE have been associated with passive transfer of immunoglobulin and persistent infection has been induced experimentally by passive transfer of antibody (146). Therefore, antibody may contribute to the establishment and maintenance of persistent nervous system infection.
The mononuclear inflammatory response in the brain includes CD4 and CD8 T cells as well as monocytes and immunoglobulin-secreting B cells (103,136,147,148). Expression of class I and class II major histocompatibility antigens is increased in brain, and β2-microglobulin, interleukin (IL)-1, soluble intercellular adhesion molecule (ICAM), soluble IL-2 receptor, and soluble CD8 are increased in CSF (149,150). Thus, there is no evidence for a global defect in immune responses, although MeV induction of interferon-γ (IFN-γ) is reduced in some individuals (151). It is likely that SSPE and MIBE are similar in their pathogenesis, but that MeV replication and the appearance of neurologic symptoms is slowed in SSPE by the presence of a vigorous immune response (64). However, this immune response is not able to clear the virus once parenchymal CNS infection has been established and infection is progressive.
Clinical Manifestations
The age of initial MeV infection in individuals subsequently developing SSPE is often younger than 2 years. The age of onset of neurologic disease is typically between 2 and 20 years of age but has been reported up to the age of 35 years (59,68,102,109). The onset is insidious and the diagnosis is often not suspected early in disease (152,153). The first symptoms are likely to be deterioration in school or work performance and changes in personality (stage I). In adults, visual impairment is often an early sign (112,154). Alteration in mental status is followed by the onset of myoclonus, convulsions, abnormal postures and movements, and autonomic dysfunction (stage II). Progressive neurologic deterioration is marked by rigidity (stage III), optic atrophy, and akinetic mutism, ending in coma (stage IV) and death months to years after onset (155–157). The course of SSPE is usually 1 to 3 years. More rapid progression has been reported with perinatally acquired infection (102,158,159). Occasionally, with good supportive care, patients survive for longer periods of time and remissions lasting weeks to years with total clinical courses of 10 to 16 years have been reported (119).
Diagnosis
The diagnosis of SSPE can be made by demonstration of high levels of antibody to MeV in CSF in the setting of a characteristic clinical picture. Typical EEG changes include a burst-suppression pattern often most easily demonstrable during sleep (94,156,160), but this may not be present in adult-onset disease (112). The CSF is often normal on routine analysis of pressure, protein, glucose, and cells. However, the immunoglobulin concentration is usually elevated, oligoclonal bands are present on electrophoretic analysis, and MeV-specific antibody is elevated (155). MeV RNA can be detected in the brain and CSF by RT-PCR (112,161,162). CT and MRI scans are generally unhelpful. CT scans often show evidence of loss of parenchyma with ventricular dilation and cortical, brainstem, and cerebellar atrophy accompanied by low parenchymal attenuation (163). MRI scans tend to show hyperintense T2-weighted lesions in gray and white matter, with white matter lesions becoming more prominent as disease progresses (112,164–166).
Treatment and Prevention
Numerous therapeutic agents including bromodeoxyuridine, azaguanine, amantadine, interferon, isoprinosine, ribavirin, and cimetidine have been used for the treatment of SSPE (162,167–170). Evaluation of the efficacy of any of these regimens is difficult because the disease is rare, most reports are anecdotal or uncontrolled, and the benefits at best are short term. Therefore, there is no established treatment for this disease.
Timely measles immunization is the most effective means of prevention. The incidence of SSPE has decreased dramatically since introduction of the live attenuated measles vaccine and there is no evidence that the vaccine virus can cause SSPE (70,101,102,104,160,171–173).
ACUTE DISSEMINATED ENCEPHALOMYELITIS
History
In 1790, James Lucas (174), an English surgeon, described the first case of postmeasles encephalomyelitis in a 23-year-old woman who developed paraparesis and urinary retention as the measles rash was fading. ADEM can develop after a number of infections and is known under many names including postinfectious and parainfectious encephalomyelitis (175). Acute hemorrhagic leukoencephalitis or acute hemorrhagic necrotizing encephalopathy probably represents a more severe form of the disease. In 1960, Koprowski (176) hypothesized that ADEM was an autoimmune (hyperergic) disease based on the similarity of the pathology and clinical picture to experimental autoimmune encephalomyelitis (EAE) and the demyelinating disease that complicates immunization with the brain-derived Semple rabies vaccine. This remains the leading hypothesis (55), but understanding of the pathogenesis of ADEM is incomplete.
Epidemiology
ADEM occurs worldwide and complicates many infections but is most frequent after measles (105,177). The overall incidence of ADEM associated with measles is approximately 1 per 1,000 infections (52) but varies with age. ADEM is more frequent in children older than 5 years of age and rare in those younger than 2 years of age (52). It occurs equally in males and females.
Pathogenesis and Pathology
ADEM occurs in close temporal association with acute measles (Fig. 8.2) at a time of an active immune response to the infection (178). Brain pathology shows perivascular inflammation and perivenular demyelination. During the acute phase, there is patchy swelling in the walls of small cerebral vessels and mononuclear cell infiltration. Later, perivenous inflammation is more marked and demyelination is evident (60,179,180).
There is little evidence for direct infection of the brain by MeV. Virus has rarely been recovered from the brain (56,181), viral antigen is not detectable by immunocytochemical staining (21,182), and viral RNA has not been detected by in situ hybridization (21). Furthermore, there is no evidence for a local virus-specific intrathecal antibody response as is expected in diseases caused by direct virus infection of the CNS (55). Therefore, there is no evidence that virus is present in the CNS at the time of neurologic disease, but more sensitive techniques such as RT-PCR have not been applied and specimens from early in disease have not generally been available for study. It is possible that virus in cerebral endothelial cells plays a role in triggering the autoimmune disease or allowing access of autoreactive cells (22). Thus, virus may no longer be detectable by the methods used at the time that tissue has been examined.
The pattern of loss of the myelin proteins, myelin-associated glycoprotein, and myelin basic protein resembles that seen in EAE, a disease induced in animals by inoculation of myelin or myelin proteins (182). Furthermore, immunologic studies of patients with ADEM have shown the presence of cellular immune responses to myelin basic protein (55,181,183) and antibody to myelin proteins similar to that seen in animals with EAE and in humans with Semple rabies vaccine-induced encephalomyelitis (55,184–187). It is postulated that ADEM is analogous to these autoimmune diseases, but the mechanism by which an autoimmune response to myelin proteins is induced during a systemic infection is not clear. The timing of this complication suggests that immune activation associated with MeV infection may play a role in induction of ADEM. Immune activation may increase presentation of self-peptides and allow the proliferation of autoreactive clones of cells (178,188). Patients with ADEM differ from patients with uncomplicated measles by having more marked and prolonged immunologic abnormalities. In particular, IgE is more persistently elevated and soluble IL-2 receptor is lower in patients with ADEM (47,189). IL-2 is one of the cytokines elevated in plasma (190), and a similar disease has been reported as a complication of IL-2 infusion (191).
The possibility of immunologic cross reactivity between some component of myelin and a component of MeV has been explored. Limited sequence homologies have been identified but none for which biologic relevance has been shown (192). Studies of MeV-specific antibodies and T cells have shown no cross reactivity with myelin basic protein or galactocerebroside (193,194).
Clinical Manifestations
Typically, patients recovering from measles present with an abrupt onset of renewed fever and obtundation accompanied by neurologic signs and symptoms that can include meningismus, seizures, altered mental status, multifocal neurologic deficits, and coma (177,180). The onset is most often between 2 and 7 days after the appearance of the rash but occasionally predates or appears up to 3 weeks after the rash (55). The disease has a monophasic course over 10 to 20 days. Improvement is usually evident within a few days after onset. Mortality ranges from 10% to 40% with substantial neurologic residua in the majority of survivors (55,60,179). Prior to widespread measles immunization, ADEM was a common cause of chronic neurologic disability.
Diagnosis
The diagnosis is apparent on clinical grounds when neurologic disease follows shortly after the rash. Routine laboratory tests are not particularly helpful. There are no consistent abnormalities in blood or urine. CSF may be normal or contain a modest elevation in protein and a mononuclear pleocytosis. The EEG shows a nonspecific diffuse slowing. The CT scan may be normal, but the MRI scan usually shows multiple foci of demyelination most likely to be seen on T2-weighted and fluid-attenuated inversion recovery (FLAIR) images in subcortical and central white matter of the cerebral hemispheres, cerebellum, brainstem, and spinal cord (166,177,195).
Treatment and Prevention
Measles vaccine is highly effective in prevention of ADEM and neurologic complications after immunization are rare (196). Treatment is not well established. Corticosteroids are widely used, but the benefit is not clear (177,197–199). One randomized study showed no benefit (200) and a retrospective study found higher mortality and rates of sequelae in steroid-treated patients (201). Intravenous immunoglobulin and plasma exchange have also been used with some reported success (177,202).
BACKGROUND ON RUBELLA
RV, the etiologic agent of the mild rash disease rubella or German measles, is the only member of the Rubivirus genus of the Togaviridae family. Togaviruses are single-stranded, positive-sense, enveloped RNA viruses and all, except RV, are in the Alphavirus genus and are transmitted by mosquitoes. No related animal viruses have been recognized. RV has three structural proteins. The capsid protein surrounds the RNA and the two envelope surface glycoproteins E1 and E2 are important for attachment and entry. E1 induces neutralizing antibodies. Two clades and 10 genotypes are recognized (203).
Like MeV, RV is a human virus that is transmitted between individuals by respiratory droplets or aerosol and spreads through the blood via a cell-associated viremia to skin and other organs (204). RV can infect many types of cells, and myelin oligodendrocyte glycoprotein has recently been identified as a cellular receptor (205). The immune response is associated with the appearance of the rash. There is substantial evidence of immune activation, leukopenia, and immune suppression, as indicated by loss of skin test responses to recall antigens (206,207).
The neurologic complications of postnatally acquired rubella include ADEM, which is in every way similar to ADEM complicating measles but occurs less frequently (approximately 1:5,000 to 8,000 cases) (208), and PRP, a disease similar to SSPE. However, the most important neurologic complications occur when RV infection is acquired prenatally (Table 8.2).

Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


