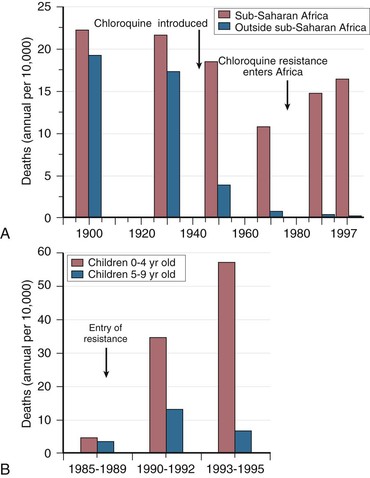
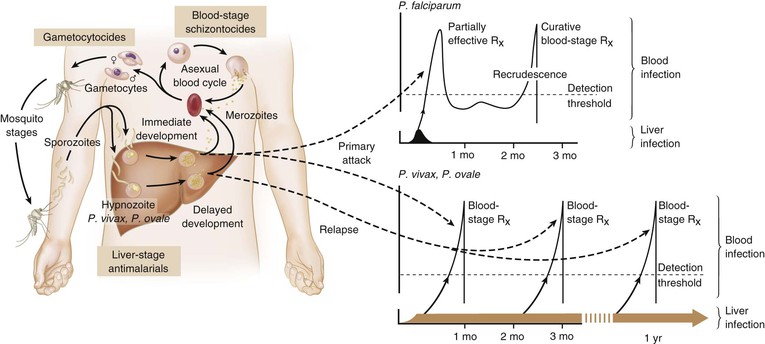
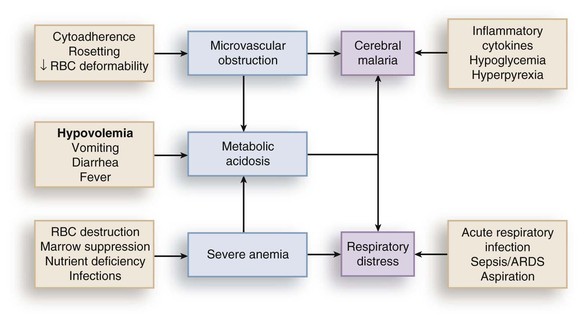
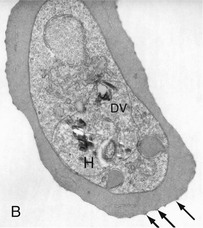
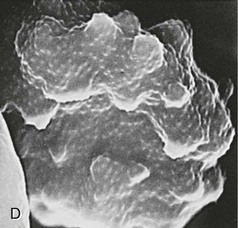
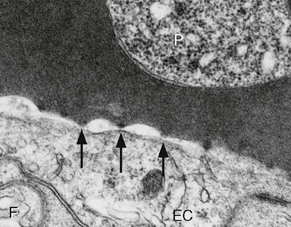
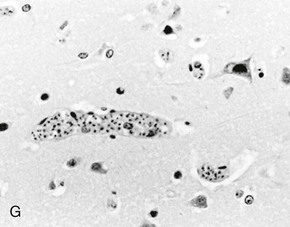
Rick M. Fairhurst *, Thomas E. Wellems Malaria remains an overwhelming problem in tropical developing countries, accounting for more than 200 million cases and more than 600,000 deaths each year.1,2,3 Nearly 40% of the world’s population is at risk for acquiring malaria.4–7 In sub-Saharan Africa, most severe cases and deaths occur in children younger than 5 years and in pregnant women. The introduction of chloroquine and dichlorodiphenyltrichloroethane (DDT) at the end of World War II brought dramatic new power to malaria control efforts worldwide. With postwar economic recovery and a renewed spirit of international cooperation, optimism ran high that the widespread use of these new compounds would eliminate malaria, and in 1955, the World Health Organization (WHO) launched its campaign to eradicate the disease. This goal proved overly optimistic, and the centrally organized DDT spraying programs at the core of the campaign were discontinued in 1967. The campaign, nevertheless, brought regional successes that coincided with other factors to reduce malaria incidence in many areas of the world (e.g., in Asia) (Fig. 276-1A).8 A stark exception to this general progress is sub-Saharan Africa, where malaria remains deeply entrenched. Even the most committed spraying and eradication programs in endemic areas of this region could not defeat malaria’s efficient transmission by the Anopheles gambiae mosquito.9 The wide availability and use of chloroquine did, however, boost the health of young African children who suffer most from Plasmodium falciparum, the species responsible for the deadliest forms of malaria. As chloroquine became increasingly available in the 1950s to 1970s, death rates from malaria in Africa began to drop, approaching half the level of the prechloroquine years.10 Unfortunately, the massive use of chloroquine (hundreds of tons sufficient for hundreds of millions of treatments annually) in the 1980s11 selected for chloroquine-resistant P. falciparum strains in Southeast Asia that entered and spread across Africa. In the 1980s and 1990s, malaria resurged and death rates increased. The impact of chloroquine resistance was especially evident in young children, who do not have the partially protective antimalarial immunity that usually develops after repeated episodes of the illness (Fig. 276-1B).12,13 In the absence of a fully effective vaccine, success against malaria in Africa will continue to depend on effective drugs, such as artemisinin-based combination therapies, that are reliable, affordable, and readily available. Of importance, increased international support and funding for prevention, control, and elimination have reinvigorated the efforts of malaria control programs in recent years, averting an estimated 274 million cases and 1.1 million more deaths between 2001 and 2010.2 Plasmodium parasites belong to the Apicomplexa group of protozoa, which includes other pathogens, such as Babesia, Toxoplasma, and Cryptosporidium species. Apicomplexa are distinguished morphologically by the presence of a specialized complex of apical organelles (i.e., micronemes, rhoptries, and dense granules) involved in host cell invasion (see Fig. 276-4A).14 Four Plasmodium species are classified as human malaria parasites: P. falciparum, P. vivax, P. ovale, and P. malariae. Some malaria parasites of other primates (e.g., P. knowlesi, P. cynomolgi, and P. simium) can also infect humans under natural conditions.15 Indeed, with the recently appreciated extent of human infections from P. knowlesi, a natural pathogen of macaque monkeys, this parasite has been proposed to be a “fifth human malaria parasite” responsible for significant morbidity and mortality in Malaysia.16–20 In 1880, Alphonse Laveran21 first observed malaria parasites in a human blood sample, witnessing the exflagellation of microgametes that usually emerge in the mosquito. It was eventually established that parasites in the bloodstream reproduce asexually in the haploid state (Fig. 276-2). During erythrocytic development, a small minority of parasites undergo a poorly understood switch to sexual-stage development. The resulting male and female gametocytes are the forms that are taken up by and infect anopheline mosquitoes, as proved by Ronald Ross and Battista Grassi22,23 in the 1890s. Gametocytes emerge from erythrocytes in the mosquito midgut as male and female gametes that cross-fertilize to form diploid zygotes, which in turn differentiate into ookinetes that burrow through the midgut wall. Each ookinete develops into an oocyst containing up to 1000 sporozoites, which emerge and are then carried by the insect hemolymph to invade the salivary glands. These processes in the mosquito require an incubation period of about 1 to 2 weeks. Female mosquitoes inject sporozoites into humans while probing the dermis in preparation for taking a blood meal. Shortt and Garnham24 demonstrated in 1948 that sporozoites must first invade and replicate in hepatocytes before they can differentiate into merozoites capable of entering the intraerythrocytic cycle. The injected sporozoites typically take several hours to travel through dermal tissues and migrate across host cell barriers before they enter blood and lymphatic systems and are carried to the liver.25 A molecular motor installed between the sporozoite plasma membrane and a double, inner membrane complex powers motility in this journey, whereas sporozoite surface proteins that are linked to this motor provide traction for gliding and crossing cellular barriers in tissue transit and invasion.26,27 Invasion of sporozoites into hepatocytes takes place by a coordinated series of steps, including host cell contact, signaling events with discharge of calcium, release of ligands and processing molecules from apical organelles, and active entry of the sporozoite into an induced parasitophorous vacuole in the hepatocyte cytoplasm. The host tetraspanin molecule CD81 is important for sporozoite entry into hepatocytes.28,29 Two recent studies have shown that the class B type 1 scavenger receptor SR-B1, a known co-receptor with CD81 for invasion of hepatocytes by hepatitis C virus (HCV), also promotes efficient Plasmodium infection of hepatocytes.30,31 A likely role for SR-B1 is the organization of CD81 into tetraspanin-enriched microdomains that are preferred membrane areas for sporozoite entry.30 Because SR-B1 is vital in providing cholesterol to the hepatocyte by high-density lipoprotein (HDL)-cholesteryl ester uptake, its exploitation by Plasmodium and HCV represents the evolutionary selection of a dependable invasion pathway by these pathogens. SR-B1’s role in HDL-cholesteryl ester uptake and activation of the liver fatty acid carrier L-FABP also supports the transformation and growth requirements of the parasite inside the hepatocyte. Individual infected hepatocytes support the development of 10,000 to 30,000 merozoites, a process that is not associated with symptoms. All P. falciparum and P. malariae parasites complete their liver-stage development in about 1 to 2 weeks.32 P. vivax and P. ovale liver stages also can develop promptly or can remain latent as hypnozoites in the liver for months to years before emerging to produce relapses of malaria (see Fig. 276-2). Once a merozoite33 egresses by protease activity from its host hepatocyte (or from its host erythrocyte in the bloodstream cycle),34 it engages loosely with an uninfected erythrocyte and then reorients so that its apical end faces the cell surface.35 The merozoite then drives itself into the erythrocyte through a ring-shaped, electron-dense junction that moves from the front to back end of the merozoite by the power of an actin-myosin motor.35 An envelope of invaginated membrane surrounds the merozoite as it enters, forming the parasitophorous vacuole once invasion is complete.36 These steps of invasion are supported by cell-signaling events, energy-dependent migration, and discharge of contents from the rhoptries, micronemes, dense granules, and perhaps other apical compartments.37 In P. falciparum, invasion can be supported by multiple different interactions between parasite molecules and erythrocyte surface molecules, including glycophorins.38,39 Recent work has demonstrated the essential role of a ligand-receptor interaction between the P. falciparum RH5 (PfRH5) protein and the Ok blood group antigen, basigin.40 A number of studies have also established a dependence of P. vivax merozoites on interaction with erythrocyte Duffy antigen receptor for chemokines (DARC),41,42 but this may not be an absolute requirement considering that P. vivax infections occur in DARC-negative individuals, for example in Madagascar.43 Within erythrocytes, merozoites develop from ring forms into trophozoites and then schizonts over 48 hours (P. falciparum, P. vivax, and P. ovale) or 72 hours (P. malariae). After breaking down their host cell membrane by enzymatic digestion, 24 to 32 merozoites enter the bloodstream and are each capable of infecting a new erythrocyte. Cycles of invasion and growth in erythrocytes produce a parasite biomass that enlarges rapidly, causing fever and leading to pathologic processes, such as erythrocyte loss (anemia) and sequestration of infected erythrocytes in microvascular beds (cerebral malaria). Malaria presents as an acute febrile illness that is often but not always characterized by the classic malaria paroxysm: chills and rigors, followed by fever spikes up to 40° C (104° F), and then profuse sweating that can ultimately give way to extreme fatigue and sleep. Paroxysms last several hours, can occur with a regular periodicity coinciding with the synchronous rupture of blood schizonts, may alternate with relatively asymptomatic periods, and are associated with high levels of tumor necrosis factor (TNF).44 Paroxysms can occur in 24-hour, tertian 48-hour, or quartan 72-hour cycles, or in other more complicated patterns.45 TNF may originate from monocytes stimulated by glycosylphosphatidylinositol moieties or other substances released on schizont rupture.46,47 Malaria can be acutely malignant and painful or more indolent and asymptomatic. It predisposes African children to bacteremia48 and increases the morbidity and mortality associated with other diseases by stressing host systems and producing effects such as dehydration, anemia, and some degree of immune suppression. Malaria is tremendously debilitating and impedes economic development through its adverse effects on fertility, population growth, saving and investment, worker productivity, absenteeism, premature mortality, and medical costs.49,50 A single episode of malaria has been estimated to result in a loss of 5 to 20 working days, and an agricultural family afflicted by malaria may be up to 60% less productive than a family without malaria.51 P. falciparum malaria can be much more acute and severe than malaria caused by other Plasmodium species (Fig. 276-3). Although P. vivax can cause serious and fatal illness,52,53 by far the largest fraction of deaths directly attributable to malaria are caused by severe complications of P. falciparum infection, including cerebral malaria, severe anemia, respiratory distress, renal failure, and severe malaria of pregnancy.54–59 Important contributory factors include metabolic acidosis, hypoglycemia, and superimposed bacterial infections. Fatal P. falciparum infections are often associated with the failure of multiple organ systems. An important feature of the pathogenesis of P. falciparum is the ability of its mature trophozoite and schizont forms to sequester in the deep venous microvasculature. This sequestration is promoted by a number of processes: the adherence of infected erythrocytes to endothelial cells60–62 (Fig. 276-4F and G), rosetting—the binding of infected erythrocytes to uninfected erythrocytes62–64 (see Fig. 276-4H), reduced erythrocyte deformability65,66 (see Fig. 276-4C and D), and platelet-mediated clumping of infected erythrocytes.67,68 In malaria of pregnancy, infected erythrocytes accumulate within the proteoglycan matrix of placental intervillous spaces.69 P. falciparum–infected erythrocytes can thus accumulate throughout the body, including the heart,70 lung,71 liver,72 brain,71,73–76 kidney,77 intestine,71,73 dermis,73 bone marrow,78 and placenta.79 Uninfected erythrocytes, monocytes, platelets, and deposits of thrombin and fibrin are often found in association with these infected erythrocytes.71,74,76,77 By sequestering in microvessels, P. falciparum may avoid filtration and destruction by the spleen and thus multiply to high densities.80 The survival and propagation of parasites may be enhanced when they sequester in the low oxygen tension environment of postcapillary venules. Attachment points to endothelium have been shown by electron microscopy to be dense protrusions, termed “knobs,” on the surface of infected erythrocytes (see Fig. 276-4D and E), where antigenically variant cytoadherence proteins (P. falciparum erythrocyte membrane protein 1 [PfEMP-1]; see later) are anchored. Attachment at knobs (see Fig. 276-4F) supports cytoadherence in vitro and sequestration in vivo (see Fig. 276-4G).81,82 Under flow conditions, cytoadherence events are reminiscent of leukocyte adhesion, involving distinct phases of tethering, rolling, and stable adhesion.83 PfEMP-1 is central to malaria pathogenesis.84 PfEMP-1 is a family of antigenically variant proteins encoded by the multicopy var gene family.85 Approximately 60 different var genes are present in the haploid genome of each parasite, encoding PfEMP-1 variants with unique antigenic and cytoadherence properties.86,87 A single PfEMP-1 variant is thought to be predominantly expressed on the surface of an individual infected erythrocyte,88 whereas all others are silenced.89 Switches in expression between individual members of the var gene family occur at an estimated rate of 2% to 18% per cell per generation90,91 and produce the antigenic variation in P. falciparum populations during the course of an infection. Broods of parasites infecting a human host may express several variants in their subpopulations. The various PfEMP-1 proteins exposed on knobs have binding domains that adhere to host molecules, including CD36, intercellular adhesion molecule 1 (ICAM-1), thrombospondin, platelet endothelial cell adhesion molecule (PECAM/CD31),83,92–95 chondroitin sulfate A (CSA),69,96 and endothelial protein C receptor involved in the deadly sequestration of parasitized erythrocytes that leads to cerebral malaria.97–99 In addition to the association of cerebral malaria with subsets of PfEMP-1 variants that adhere to brain endothelium, other pathologic conditions have also been linked to particular PfEMP-1 variants and host receptors.60,100–105 CD36 is an important cytoadherence receptor expressed on microvascular endothelium, as well as on monocytes and platelets, and is thought to mediate the sequestration of parasites as well as the host immune response to infection.106 Infected erythrocytes that bind CSA expressed by syncytiotrophoblasts sequester selectively in placental tissue and are responsible for malaria of pregnancy.69 Some PfEMP-1 variants are also important parasite ligands in rosetting107 because they can adhere to complement receptor 1 (CR1)108 and blood group A antigen109 on uninfected erythrocytes. A human CR1 polymorphism that reduces P. falciparum rosetting was found in one study to protect against severe malaria110; data from other studies of rosetting and disease severity have, in some cases, shown an association111–115 and in others have not.116,117 PfEMP-1 variants that bind to glycoprotein CD36 on the surface of platelets are also thought to play an important role in the platelet-mediated clumping of infected erythrocytes.67 High parasite densities,118 increased parasite multiplication rates,119 and evidence of high parasite biomass (e.g., intraleukocytic pigment, mature trophozoites, and schizonts) on peripheral blood smears are associated with increased severity of malaria and death.120–122 P. falciparum can infect erythrocytes of all ages,123 promoting heavy parasite burdens; P. vivax selectively infects reticulocytes124,125 and thus does not achieve high densities in the peripheral blood. The classic histopathologic finding of fatal cerebral malaria is the intense sequestration of infected erythrocytes in cerebral microvessels (see Fig. 276-4G), often accompanied by “ring” hemorrhages, perivascular leukocyte infiltrates, thrombin deposition, activated platelets,75,126–128,129,130 and immunohistochemical evidence for endothelial cell activation.74,131,132 In one autopsy series of patients who died from cerebral malaria, 94% of brain microvessels contained adherent infected erythrocytes, compared with 13% of patients who died from noncerebral malaria.133 In another study, 7 of 31 (24%) African children who received a clinical diagnosis of cerebral malaria were found at autopsy to have nonmalarial causes of coma, underscoring the possibility that other illnesses may mimic the cerebral malaria presentation in areas where incidental parasitemia is common.128 Sequestration of infected erythrocytes stimulates the local production of inflammatory cytokines, such as TNF, elevated levels of which may correlate with disease severity.134–136 These cytokines and other inflammatory mediators also upregulate adhesion molecules, such as ICAM-1, in the cerebral microvasculature,137,138 which may lead to further sequestration of infected and uninfected erythrocytes, leukocytes, and activated platelets.126 Impaired nitric oxide bioavailability may also contribute to endothelial dysfunction139 by increasing microvascular tone, endothelial cell adhesion molecule expression, cytokine production, and infected erythrocyte sequestration.140 These processes may cause varying degrees of functional obstruction and consequently impair local delivery of oxygen and glucose. However, obstruction by infected erythrocytes and other blood elements does not generally produce neurologic sequelae akin to those that follow the physical occlusion in thrombotic stroke because most patients with cerebral malaria who recover can do so rapidly within 48 hours and without such consequences. Systemic sequestration of metabolically active parasites, blood cells, and platelets likely contributes to the metabolic acidosis and thrombocytopenia commonly seen in severe malaria. Metabolic acidosis, hypoglycemia, hyperpyrexia, and nonconvulsive status epilepticus can contribute significantly to the cerebral malaria presentation, as suggested by the rapid clinical improvement of some patients after fluid resuscitation, blood transfusion, dextrose infusion, fever reduction, and anticonvulsant therapy in addition to antimalarial treatment.141–144 Hypoglycemia in malaria can cause coma and convulsions and thus may contribute substantially to the morbidity and mortality associated with cerebral malaria.54 The pathophysiologic mechanisms of hypoglycemia in children and adults seem to be different. In children, insulin levels are appropriate and hypoglycemia is associated with impaired hepatic gluconeogenesis and increased consumption of glucose by hypermetabolic peripheral tissues.143,145–148 Large amounts of glucose are also consumed by intraerythrocytic parasites.149 In adults, hypoglycemia is often associated with hyperinsulinemia,150 which may result from pancreatic islet cell stimulation by parasite-derived factors and/or parenteral quinine or quinidine therapy.151 Depletion of liver glycogen stores after decreased food intake during the prodromal period may also contribute to hypoglycemia. The pathophysiology of malarial anemia is multifactorial and complex.152–154 The intravascular lysis and phagocytic removal of infected erythrocytes155 contribute to anemia but do not account for the dramatic reductions in erythrocyte mass that can accompany acute P. falciparum malaria episodes. Additional processes have therefore been implicated in the pathogenesis of malarial anemia. Excess removal of uninfected erythrocytes may account for up to 90% of erythrocyte loss156 and may be mediated by processes (e.g., oxidative stress) that accelerate the senescence and reduce the deformability of erythrocytes. The contribution of impaired erythropoietic responses to malarial anemia is significant and probably involves general processes also found in other diseases. Release of inflammatory cytokines (e.g., TNF) are associated with impaired production of erythropoietin,157,158 decreased responsiveness of erythroid progenitor cells to adequate levels of erythropoietin,159,160 and increased erythrophagocytic activity.161 These pathogenic processes account for the normochromic/normocytic anemia seen in malaria, and explain the notable absence of a robust reticulocyte response. Although microcytosis and hypochromia are seen in malaria, these findings are often attributable to thalassemias and iron deficiency in endemic areas. Bacteremia, nutritional deficits (e.g., vitamin A and B12 deficiencies), concomitant infections (e.g., hookworm and Schistosoma), and genetic polymorphisms (glucose-6-phosphate dehydrogenase [G6PD] deficiency) have also been associated with greater degrees of anemia in malaria episodes,162 presumably by lowering the baseline from which hemoglobin levels acutely decline. In endemic areas where chloroquine resistance is prevalent, the inability of young children to clear their parasitemias with chloroquine contributes to their higher baseline prevalence of anemia when compared with children treated with more effective drugs.163 The most significant pulmonary manifestation directly attributable to P. falciparum is noncardiogenic pulmonary edema.164,165 Sequestration of infected erythrocytes in the lungs is thought to initiate regional production of inflammatory cytokines that increase capillary permeability, leading sequentially to pulmonary edema, dyspnea, hypoxia, acute lung injury, and acute respiratory distress syndrome (ARDS).166,167 Pulmonary edema is common with severe malaria in adults but infrequent in children, and it is not associated with pleural effusion. Iatrogenic fluid overload and acute renal failure may contribute to the development or worsening of pulmonary edema. Although pulmonary edema usually occurs after other features of severe disease (e.g., coma, acute renal failure) become manifest, it may occur at any time during the clinical course, even when the patient appears to be recovering on antimalarial therapy. Dyspnea and increased respiratory rate are features of impending pulmonary edema and often precede other clinical (e.g., use of accessory muscles of respiration) and radiologic signs (e.g., generalized increase in interstitial markings). Pulmonary manifestations of deep breathing and respiratory distress associated with severe malaria may also arise from metabolic acidosis,55 severe acute respiratory infections,147 sepsis-related ARDS, aspiration (especially with diminished consciousness or convulsions), and nosocomial pneumonia. Cerebral pathologic processes may result in abnormal breathing patterns including Cheyne-Stokes respirations and respiratory failure.168 Metabolic acidosis is a common feature of severe malaria and is associated with significant lactic acidemia in up to 85% of cases. Metabolic acidosis is principally caused by reduced delivery of oxygen to tissues, from the combined effects of anemia (decreased oxygen-carrying capacity), sequestration (microvascular obstruction), and hypovolemia (reduced perfusion) resulting from fluid losses caused by fever, decreased oral intake, vomiting, and diarrhea.169 These effects produce a shift from aerobic to anaerobic metabolism and cause lactate levels to increase.170 The following factors may also contribute to metabolic acidosis: production of lactate by anaerobic glycolysis in sequestered parasites;171 reduction of hepatic blood flow, leading to diminished lactate clearance;172,173 induction of lactate production by TNF and other proinflammatory cytokines;174 impairment of renal function;172 and ingestion of exogenous acids (e.g., salicylate) or unknown constituents of traditional herbal remedies for fever.175 Placental malaria results in maternal morbidity and mortality, intrauterine growth retardation, premature delivery, low birth weight, and increased newborn mortality.176–178 Selective accumulation of infected erythrocytes in the placenta involves their interaction with syncytiotrophoblastic CSA,69 which is possibly complemented by interactions with other molecules, such as hyaluronic acid and immunoglobulins.179–181 This is in contrast to the sequestration of infected erythrocytes in the systemic microvasculature, where CD36 is the major endothelial receptor. Parasites that accumulate in the placenta express PfEMP-1 variants that bind CSA182,183 but not CD36.184 Evidence suggests that women experiencing a malaria episode from CSA-binding parasites during their first pregnancy lack immunity to the PfEMP-1 antigenic variants presented by these strains and, despite immunity to CD36-binding variants from previous infections, are highly susceptible to the new placental infection. Placental malaria in subsequent pregnancies is typically less severe than in the first pregnancy,185 presumably because of a woman’s previous experience with CSA-binding parasites. Infections with P. vivax and P. ovale can be considered similar to each other from a clinical perspective. P. vivax infections can be tremendously debilitating and are sometimes associated with serious complications, including acute lung injury186,187 and splenic pathologies.188,189 Splenic rupture has been associated with acute and chronic infections and can occur spontaneously or with minor trauma, including manual examination of the spleen. Although more commonly associated with P. vivax malaria, splenic rupture has been associated with all four human malaria parasites. Anemia is frequently observed as a consequence of acute or chronic infections, or as a result of repeated acute infections.155 Suppressed erythrocyte production and hemolysis of both infected and uninfected erythrocytes have been implicated in the pathogenesis of P. vivax malarial anemia. Although not often fatal, P. vivax infections have recently been associated in Papua New Guinea and Indonesia with severe disease manifestations, including cerebral malaria, severe anemia, and respiratory distress.52,53,190 P. vivax merozoites selectively invade reticulocytes.125 Because these cells account for only a small proportion of the total erythrocyte mass, parasitemias in P. vivax infections are usually less than 1%. Recent evidence suggests that P. vivax can cytoadhere to lung endothelial cells191 and can cause lung injury by sequestering in the pulmonary microvasculature.192 P. vivax parasites may avoid splenic entrapment by increasing rather than decreasing erythrocyte deformability.193
Malaria (Plasmodium Species)
The Malaria Problem
Plasmodium and Its Life Cycle
Pathophysiology
The Malaria Paroxysm and General Considerations
Plasmodium falciparum
Cerebral Malaria
Hypoglycemia
Anemia
Pulmonary Edema and Respiratory Distress
Metabolic (Lactic) Acidosis
Malaria of Pregnancy
Plasmodium vivax and Plasmodium ovale
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Malaria (Plasmodium Species)
276