INTRODUCTION
SUMMARY
Lymphopoiesis refers to the process by which the cellular components of the immune system (i.e., T cells, B cells, and natural killer cells, and certain dendritic cells) are produced during hematopoietic differentiation. This process begins with the hematopoietic stem cell and continues through progenitor stages down a series of mostly diverging lineage pathways, ultimately resulting in the remarkable diversity and flexibility of the immune system. Although the more terminal events in lymphocyte differentiation and function have been defined in detail (Chaps. 75 to 77), the earliest events during which hematopoietic stem cells undergo lymphoid lineage commitment are less-well understood and still controversial. Although the conceptual framework for the questions of lymphoid commitment has been established largely on studies in the mouse, experimental systems now exist to better understand how such events are controlled in humans. This chapter summarizes what is known about the ontogeny of lymphoid development and the control of lymphoid differentiation, and discusses some of the persisting controversies in the field.
Acronyms and Abbreviations
AGM, aorto-gonad-mesonephros; BM, bone marrow; BCR, B-cell receptor; CLP, common lymphoid progenitor; CT, computed tomography; DC, dendritic cell; DN, double negative; E, days of gestation; EBF, early B-cell factor; FACS, fluorescence-activated cell sorting; FLT3, Fms-like tyrosine kinase 3; HSC, hematopoietic stem cell; Ig, immunoglobulin; IL, interleukin; JAK3, Janus kinase 3; LMPP, lymphoid-primed multipotent progenitor; LSK, linnegsca-1+c-kit+; NK, natural killer; PAS, para-aortic splanchnopleura; SCID, severe combined immunodeficiency.
LYMPHOPOIESIS DURING PRENATAL DEVELOPMENT
Blood is formed from a succession of sites during embryonic and fetal development, beginning outside the embryo in the yolk sac. Soon afterward, hematopoiesis begins in the embryo proper, initially in the para-aortic splanchnopleura (PAS) and aorto-gonad-mesonephros (AGM) regions, then the fetal liver, spleen, and finally the fetal marrow (Chap. 7). With each change of anatomical site, the range of hematopoiesis becomes progressively more complex and similar to that of the adult (Fig. 74–1).
Figure 74–1.
Timing of lymphohematopoiesis during prenatal development. Shown is the timeline for activity in each site of hematopoiesis in the embryo and fetus of (A) human and (B) mouse. B and T cells are first detected in vivo in fetal liver and thymus, respectively, at times shown. AGM, aorto-gonad-mesonephros; PAS, para-aortic splanchnopleura.
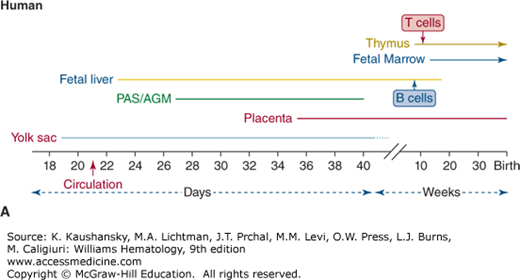
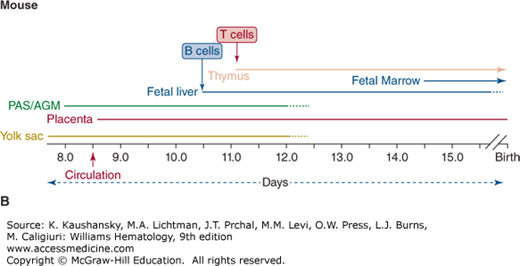
When assigning hematopoietic function to each developmental stage, it is important to distinguish the lineage “potential” of stem and progenitor cells that arise from certain areas (i.e., the ability to generate specific lineages in vitro from immature cells removed from a region) from the spontaneous physiologic production of lineages in each region. With this distinction in mind, the onset of lymphopoiesis during embryogenesis lags behind development of the myeloid and erythroid lineages. Although myeloid, erythroid, and natural killer (NK) cells can be produced from all extraembryonic and embryonic sites, B and T lymphocytes are predominantly generated from so-called definitive hematopoietic stem cells (HSCs) in the embryo proper.1
Most of the studies exploring embryonic and fetal hematopoiesis have been performed using mouse models. Although the timing of each developmental stage has been carefully mapped, it has long been a source of controversy as to whether hematopoiesis in the embryo is initiated from colonizing precursors from the extraembryonic yolk sac, or whether the embryonic sites of hematopoiesis arise independently from the yolk sac.2,3,4,5,6 This debate has implications for understanding the lineages generated at different sites of hematopoiesis and thus for tracing the ancestry of the lymphoid cells that are produced in the mammalian embryo. One reason for the difficulty in assigning the exact organ in which lineages are generated, is that each site of hematopoiesis is active during overlapping periods (see Fig. 74–1). In addition, once circulation has been established, it is difficult to rule out the possibility that stem cells and progenitors found in one location did not migrate from another. However studies using Ncx1–/– mice, which lack both heartbeat and circulation,7,8 are beginning to dissect the autonomous lineage potentials of these distinct embryonic hematopoietic tissues.
The first wave of hematopoiesis in the mouse begins in the extraembryonic tissue of the yolk sac by 7.5 days of gestation (E7.5), before circulation is established.9,10 This initial stage of so-called primitive hematopoiesis produces mostly erythrocytes and macrophages. Although lymphocytes are not detectable at this time,10 the contribution of first-wave progenitors to downstream fetal lymphopoiesis has been suggested by the identification of a lymphomyeloid progenitor in the E9.5 yolk sac, which expresses Rag-1, one of the earliest lymphoid-specific events,11 as well as of a distinct progenitor with B-1/marginal zone B cell potential.12 Further studies have identified both thymic-repopulating and multipotent potential in the yolk sac,13,14 indicating emerging changes to our understanding of primitive hematopoiesis. The murine placenta has also been identified as an autonomous source of multipotent hematopoietic cells as early as E8.58; however, the direct contribution of either yolk sac or placental progenitors to definitive lymphoid development remains to be determined. Definitive HSCs that are capable of generating all lymphohematopoietic lineages first appear in the PAS/AGM region at E8.5 to E9.3,10 High-level, multilineage reconstituting activity typical of definitive HSC can be found in the murine AGM region by E10.5. However, although AGM cells can produce all lineages, including T and B lymphocytes in vitro, lymphocytes do not spontaneously develop in the fetus until hematopoiesis has begun in the fetal liver. Rag-1 expression, one of the earliest lymphoid-specific events, can be found in the E11 murine fetal liver.10 T-cell potential has been identified in the yolk sac and PAS as early as E8.25 to E9.5 of murine gestation13; however, T-cell differentiation in vivo begins with the colonization of the thymus around E11 by stem or progenitor cells that migrate to the thymus from the AGM, fetal liver, and, later still, the fetal marrow.15,16
Hematopoietic cells have been identified in the human yolk sac as early as day 18 of embryonic life, at which time, like the mouse, they are almost exclusively comprised of erythrocytes and, to a lesser extent, monocytes and macrophages (see Fig. 74–1).17 Although no lymphocytes are seen in the yolk sac, yolk sac progenitors do have NK cell potential under certain in vitro conditions.17,18 The same yolk sac progenitors, however, do not possess the capacity for B- or T-cell development, even when placed in culture conditions that permit lymphoid differentiation.18
As in the mouse, definitive hematopoiesis develops first in the AGM region derived from the splanchnopleura, as evidenced by the finding that the AGM is the site where CD34+ cells with the capacity for full lymphoid and myeloid differentiation are first found in the human embryo.18,19 The AGM develops at day 27 of gestation in the human, when human HSCs are generated as clusters of two or three cells arising from the endothelium specifically on the ventral surface of the preumbilical region of the aorta. These cells are clonogenic and highly proliferative, rapidly increasing to several thousand in number and spreading further along the aorta. However, hematopoiesis exists only transiently in the AGM, disappearing entirely by day 40.17 Although lymphoid cells can be produced in culture from cells extracted from the AGM17,18 the HSC of the AGM do not produce mature cells in situ; instead their role is to migrate and colonize the fetal liver, producing the next wave of hematopoiesis. As in the mouse, recent studies have also detected multipotent progenitors with lymphoid potential in the human placenta. These reports suggest that hematopoietic stem/progenitor cells may also be generated de novo from the large vessels of the chorionic plate as early as week 5 of gestation, with multipotent HSC detectable at 15 weeks.20,21 However, the contribution of placental HSC to fetal liver or marrow colonization remains unclear.
Although blood cells are first detectable in the human fetal liver as early as day 23, they exist at this time only as erythroid and myeloid cells associated with hepatic sinusoids. These erythroid cells consist of megaloblasts expressing embryonic hemoglobins (globin chains ζ and ε), and no CD34+ cells are seen in the fetal liver during this early phase. It is likely that this first stage of fetal liver hematopoiesis is secondary to colonization of more mature cells from the yolk sac. By day 30, AGM-derived CD34+ cells appear in the fetal liver,22 and by day 32, these cells are able to maintain long-term hematopoiesis in vitro.22 Erythroid cells in the fetal liver at this later stage of definitive hematopoiesis consist of enucleated macrocytes producing fetal hemoglobin (globin chains α and γ). In normal development, as with the yolk sac and AGM, hematopoiesis in the fetal liver is transient, disappearing by 20 weeks of gestation.1
The final wave of hematopoietic development takes place in the fetal marrow, starting around 11 weeks of gestation. The initial cells seen in the marrow are CD15+ myeloid cells and glycophorin A+ erythroid cells, and hematopoiesis is again associated with the endothelium, taking place in the medullary sinusoids before osteoblast formation.23 Eventually, CD34+ cells are found in the fetal marrow and behave functionally as true HSC, generating B, T, NK, and myeloid and erythroid lineages.1 HSC have found their final niche, and lifelong, self-renewing lymphohematopoiesis resides permanently in the marrow thereafter.
The human thymic microenvironment begins to develop at approximately 4 weeks’ gestation and then undergoes three developmental phases.24 The first phase occurs between 4 and 8 weeks’ gestation, with the appearance of thymic epithelium arising as a product of the third and fourth pharyngeal pouches25 and the expansion of thymic epithelial cells. The second phase occurs between 9 and 15 weeks’ gestation and is characterized by the development of subcapsular, cortical, and medullary regions.24 Thymic colonization by fetal liver-derived progenitors and lymphocyte production begins at approximately week 9.25 The ability of thymocytes to respond to the mitogen phytohemagglutinin is detectable as early as 10 weeks’ gestation,26 and alloreactive, phenotypically mature T cells can be found by 13 to 16 weeks’ gestation.27
The third phase occurs from 16 weeks’ gestation until age 1 to 2 years and is characterized by robust intrathymic T-cell maturation (Chaps. 6 and 76).
An exhaustive study of 136 human postnatal thymuses ranging from neonatal life to more than 90 years old, found that essentially all postnatal thymic growth (based on weight and volume) occurs during the first postnatal year, mostly in the first few months of life.28 From the age of 12 months, the human thymus undergoes steady involution, with a reduction of thymocytes and thymic epithelium, particularly the medulla, and a corresponding increase in fatty infiltration of the perivascular space.28
Whereas mice lose approximately 90 percent of the wet weight of the thymus during life, the increasing fatty infiltration in the perivascular space in the aging human thymus maintains the total size of the thymus in healthy people into late life.28 Radiologic studies using computed tomography (CT) scans have confirmed that total thymic size remains stable in humans throughout life although parenchymal tissue atrophies dramatically (~95 percent; Chaps. 6 and 9).29
The hallmark characteristic of a mature B cell circulating in blood or residing in secondary lymphoid tissue is the expression of cell-surface immunoglobulin (Ig). The cell-surface Ig consists of μ, δ, γ, α, or ε heavy chains disulfide-linked to κ or λ light chains (Chap. 75). The cell-surface Ig and the associated signaling molecules Igα (CD79a) and Igβ (CD79b) are referred to as the B-cell receptor (BCR). Progenitor (pro-) B cells are defined by the absence of both cytoplasmic μ heavy chains and cell-surface BCR. Precursor (pre-) B cells are defined by the presence of cytoplasmic μ heavy chains in the absence of cell-surface BCR. This minimal definition of pro-B, pre-B, and B cells forms the basis of the current detailed model of human B-cell development.30 B-cell development can be divided into two stages: an antigen-independent stage that occurs primarily in fetal liver and fetal and adult marrow, and an antigen-dependent stage that occurs primarily in secondary lymphoid tissue, such as spleen and lymph node.
The first B cells detectable in the human fetus are found in the fetal liver25 at approximately 8 weeks’ gestation, with the appearance of cytoplasmic IgM+ pre-B cells; by 10 to 12 weeks, surface IgM+ B cells are seen in the fetal liver31 and fetal omentum.32 B-cell and IgM production move to the fetal marrow and spleen by 17 weeks of gestation (Chaps. 6, 7, and 75).33,34 From the end of the second trimester throughout adult life, marrow is the exclusive origin of B-cell development.35 The frequency of early B-lineage cells as a percentage of the total nucleated lymphohematopoietic cell pool is higher in fetal than in adult marrow. However, the ratio between pro-B, pre-B, and immature B cells and the mitotic activity within these fractions is relatively constant.36
In murine B-cell development, two functionally and immunophenotypically distinct types of B cells, B-1 and B-2, have been described.31 Most B cells in adult mice are B-2 cells, which form part of the adaptive immune system by their ability to interact with T cells and undergo immunoglobulin heavy chain class switching. The B-1 cells make up approximately 5 percent of adult murine lymphocytes, but demonstrate a far less diverse immunoglobulin repertoire than the B-2 cells, responding to carbohydrate antigens and other T-cell–independent immunogens and forming part of the innate immune system. Murine B-1 cells are marked by their expression of CD11b, and are found in multiple sites, including the spleen, intestine, and the pleural and peritoneal cavities.37,38 The B-1 cells can be further divided into B-1a cells (which secrete immunoglobulins spontaneously) and B-1b cells (in which immunoglobulin production is induced) based on the expression of the marker CD5. The demonstration of a B-1a/marginal zone B-cell–restricted progenitor in the E9 extraembryonic yolk sac suggests the B-1 lineage may emerge prior to the development of the adaptive immune system.12 At present, there is no clear evidence that humans have similar subpopulations of B-1 and B-2 cells during development.31
Functional NK cells can be detected in the human fetal liver as early as 9 to 10 weeks of gestation,26 but NK cell differentiation can be induced in vitro from progenitors derived at all stages of hematopoietic development, even those from the yolk sac.1,17,18 Thus, the onset of NK potential is not equivalent to the full lymphoid potential of definitive hematopoiesis, as NK potential can be assigned to a range of progenitor types that exist at different stages, including primitive hematopoiesis. NK cell production can be considered as providing an essential defense mechanism for the developing mammalian embryo prior to development of more complex pathways of adaptive immunity.
Dendritic-like cells which express class II major histocompatibility (MHC) antigens are produced at all stages of embryonic and fetal hematopoiesis, being first detected in the human yolk sac and mesenchyme as early as 4 to 8 weeks, before development of the fetal marrow or thymus.31 Dendritic cells (DCs) are detectable at each site of hematopoiesis as soon as they become active, in the human fetal thymus at 11 to 14 weeks, marrow at 14 to 17 weeks, spleen at 16 weeks, and tonsils at 23 weeks.39,40 DC and macrophages are closely related and phenotypically similar (Chap. 67), both expressing MHC class II. Thus, clear discrimination of these two cell types can be difficult, particularly as many studies that provided information on human DC development were conducted before all the molecular and antibody tools for analysis of DC were available.
DIFFERENTIATION PATHWAYS FOR LYMPHOCYTE PRODUCTION
The conceptual framework for how the lymphocyte lineages are generated from HSC was developed largely from studies using genetically engineered mice and murine transplant models. Although necessary and useful as a starting point, caution should be exercised in translating the results of the murine studies to human lymphopoiesis, or in assuming for any species that only one pathway to lymphopoiesis exists at all stages of ontogeny.41,42 Additionally, the conclusions about lineage relationships of isolated populations are influenced by limitations inherent in any of the in vitro or in vivo assays employed to examine differentiation potential.43
For several decades, our understanding of hematopoiesis has been built on a hierarchical schema in which all the pathways of differentiation lead away from a multipotent HSC, and progress through discrete progenitor stages that mark each branch-point of lineage commitment (Chap. 18). In the classical paradigm, the earliest differentiation “decision” made by an HSC is to enter one of two pathways, marked by either a common lymphoid progenitor (CLP) or a common myeloid progenitor (CMP), which has full myeloid and erythromegakaryocytic differentiation potential (Fig. 74–2).44 With each successive stage of differentiation, lineage-specific cell-surface markers and transcription factors are upregulated, and alternative lineage potentials are lost. Consequently, the CLP is defined as a single cell that can give rise to all lymphoid lineages (B, T, and NK), but cannot generate myeloid, erythroid, or megakaryocytic lineages. The concept of progenitor populations marking two mutually exclusive differentiation pathways, one limited to myeloid and erythromegakaryocytic potential and the second defining lymphoid commitment, was held long before cells that satisfied the criteria of CLP were identified.45,46,47 In contrast, the existence of single clonogenic cells with myeloid, erythroid, and megakaryocyte lineages was shown more than three decades ago through the use of in vitro clonal assays to demonstrate so-called colony-forming unit-granulocyte erythromyeloid megakaryocyte,48 and later confirmed using markers to prospectively isolate such cells in mice49 and humans.50 The lymphocyte lineages were assumed to be closely related because of a number of associations, for example, common anatomical sites of T and B lymphopoiesis (spleen, lymph nodes; Chap. 6), similar molecular mechanisms that regulate T-cell receptor and B-cell immunoglobulin rearrangements (Chaps. 75 and 76), and severe B- and T-lymphoid defects that result from single genetic mutations in mice (Chap. 80).51
Figure 74–2.
Postnatal pathways of lymphopoiesis in mice and humans. The key immunophenotype used to isolate each population is shown in boxes on the right. In parentheses under the main immunophenotypes are other markers also associated with each population. The exact relationship between lymphoid-primed multipotential progenitors (LMPPs) and common lymphoid progenitors (CLPs), and between LMPPs and myeloid progenitors remains controversial, as does the main cell type that initiates thymopoiesis (all shown as dashed lines). In both mice and humans, dendritic cells (DCs) can be produced in vitro from all prospectively identified lymphoid progenitors as well as myeloid progenitors. A. Murine lymphoid progenitor pathways. Populations with multilineage potential include long-term hematopoietic stem cells (LT-HSCs) and multipotential progenitors (MPPs). FLT3+ LMPPs, have full lymphoid (T-, B-, and natural killer [NK] cell developmental potential) and limited myeloid (nonclonogenic, mostly monocyte) potential. In the CLP, all myeloid potential is lost and full lymphoid potential remains. B. Human lymphoid progenitor pathways. Populations with multilineage potential include HSC and LMPP (aka multilymphoid progenitor [MLP]), which have full lymphoid (T-, B-, and NK cell developmental potential) and limited myeloid (mostly monocyte) potential. The CD10+ CLP has predominantly B, and some NK and T potential and is almost devoid of myeloid potential. Alternative phenotypic markers reported for each progenitor type with similar lineage potential are shown. BM, marrow; GMP, granulocyte-macrophage progenitor; MEP, megakaryocyte-erythroid progenitor.
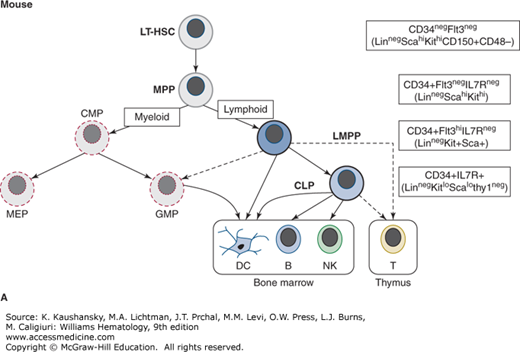
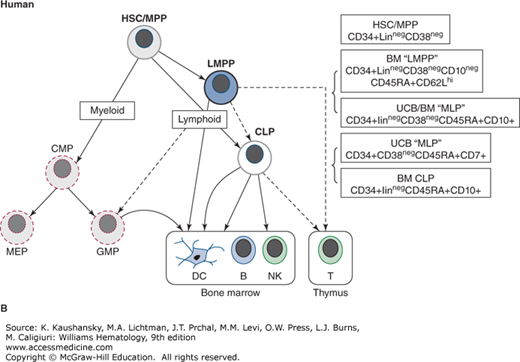
Development of flow cytometry (synonym: fluorescence-activated cell sorting [FACS]) made possible the isolation of rare hematopoietic cell populations and the subsequent interrogation of lineage potential using in vitro cultures and in vivo reconstitution studies (Chap. 18). Primitive multilymphoid progenitors with little or no clonogenic myeloid or erythroid potential have now been isolated from human tissue using flow cytometry with combinations of various cell-surface markers.52,53,54 However, it seems likely that lineage relationships are less-strictly organized than once believed. Studies in mice show that the erythroid and megakaryocytic lineages can branch off at an earlier point in hematopoiesis, and that lymphoid (i.e., T, B, and NK) and myeloid (or at least monocytic) lineages can arise from the same pathway through a so-called lymphoid-primed multipotent progenitor (LMPP).55 It remains unclear which of these lineage differentiation pathways are most physiologically significant during steady-state hematopoiesis, but it is likely that more than one pathway can exist simultaneously and alternative pathways may predominate during different stages of ontogeny and from different sites of hematopoiesis.
In 1997, investigators working with murine marrow cells, isolated progenitors that possessed no myeloid or erythromegakaryocytic potential, but when transplanted into irradiated recipients could rapidly restore T-, B-, and NK cell lineages.56 Clonal in vitro and in vivo studies showed that all lymphoid lineages were derived from a single common progenitor, thus proving the existence of the long-assumed CLP and supporting the classical model of lymphopoiesis. This study isolated cells based in part on expression of interleukin-7 receptor alpha (IL-7Rα).56 The IL-7Rα+ CLP do not express hematopoietic markers associated with fully differentiated lineages (they are called “lineage negative” or “linneg” cells). As an indication that they are more differentiated than multilineage HSCs, expression of certain HSC-related cell-surface markers (Sca-1, Thy-1, c-kit) is downregulated.56 Thus the full immunophenotype assigned to the murine CLP is Linneg IL-7Rα+ Thy-1neg Sca-1lo c-kitlo. This contrasts the murine CLP immunophenotype with that of the murine HSC, which is found within the Linneg IL-7Rαneg Thy-1lo Sca-1hi c-kithi population.56
Work with murine marrow has prompted a reexamination of when the lymphoid lineage pathways diverge from those of the myeloid and erythroid lineages (see Fig. 74–2). A population from murine marrow cells, defined largely by expression of the receptor FLT3 (Fms-like tyrosine kinase 3), has been shown to possess full lymphoid and some myeloid potential, but not erythroid or megakaryocytic potential.55,57 These linnegsca-1+c-kit+CD34+FLT3hi (synonym: LSK CD34+FLT3hi) cells are primed for lymphoid commitment in that they have downregulated genes involved in erythromegakaryocytic differentiation and upregulated lymphoid-associated genes.58
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


