INTRODUCTION
SUMMARY
Lymphocytosis is defined as an absolute lymphocyte count exceeding 4 × 109/L, whereas lymphocytopenia is defined as a total lymphocyte count less than 1.0 × 109/L. Lymphocytosis can be categorized as either polyclonal or monoclonal. Monoclonal lymphocytosis reflects an underlying clonal lymphoid disease in which the numbers of lymphocytes are increased because of the acquisition of somatic mutations resulting in clonal expansion of a lymphocyte progenitor. This expansion can be stable, such as monoclonal B-cell lymphocytosis or a progressive malignancy such as acute lymphocytic leukemia, whereas polyclonal lymphocytosis is most commonly the result of stimulation or a reaction to factors extrinsic to lymphocytes, generally infections and/or inflammation. Lymphocytopenia, on the other hand, typically reflects depletion of T cells, the most abundant lymphocyte subtype in the blood. The most common cause of such T-cell depletion is a viral infection, such as infection with the human immunodeficiency virus, although other causes exist. This chapter outlines the conditions associated with abnormalities in the numbers of circulating lymphocytes in the blood. It also serves as a useful road map to other chapters in the book that describe in detail those conditions that commonly are associated with abnormalities in the absolute numbers of circulating lymphocytes.
Acronyms and Abbreviations
BNP, brain natriuretic peptide; CLL, chronic lymphocytic leukemia; CML, chronic myelogenous leukemia; CMV, cytomegalovirus; EBV, Epstein-Barr virus; GVHD, graft-versus-host disease; IFN-γ, interferon-gamma; Ig, immunoglobulin; IL, interleukin; LCK, lymphocyte-specific kinases; LGLL, large granular lymphocytic leukemia; MBL, monoclonal B-cell lymphocytosis; miRNA, microRNA; NK, natural killer; PPBL, persistent polyclonal B-cell lymphocytosis; TCR, T-cell receptor; UNC, uncoordinated; WHO, World Health Organization.
LYMPHOCYTOSIS
Lymphocytosis is defined as an absolute lymphocyte count exceeding 4 × 109/L, although somewhat higher threshold values (e.g., >5.0 × 109/L) are sometimes used. The normal absolute lymphocyte count is significantly higher in childhood. Chapter 2 describes the methods for determining the absolute lymphocyte count and the normal range for such counts in older children and adults (see Chap. 2, Tables 2–1 and 2–2). Tables 7–3 and 7–4 in Chapter 7, provide the lymphocyte counts and lymphocyte subset counts in newborns and infants.
The blood film of patients with lymphocytosis should be evaluated for a predominance of reactive lymphocytes associated with infectious mononucleosis (Chap. 82), large granular lymphocytes associated with large granular lymphocytic leukemia (Chap. 94), smudge cells associated with chronic lymphocytic leukemia (CLL; Chap. 92), or blasts of acute lymphocytic leukemia (Chap. 91). Chapter 73 provides a description of normal lymphocyte morphology.
Characterization of cell-surface markers is valuable in distinguishing primary lymphocytosis (leukemic) from secondary lymphocytosis (reactive). Improvements in flow cytometric techniques and reagents have allowed clinical laboratories to perform flow cytometric immunophenotyping to distinguish benign from neoplastic lymphoproliferative disease.1 Analysis for immunoglobulin or T-cell receptor gene rearrangement also may provide evidence for monoclonal B-cell or T-cell proliferation, respectively.1,2
Primary lymphocytosis defines conditions associated with an increase in the absolute number of lymphocytes secondary to an intrinsic defect in the expanded lymphocyte population (Table 79–1). These conditions also are referred to as lymphoproliferative disorders and most commonly are secondary to the neoplastic accumulation of monoclonal B cells, T cells, natural killer (NK) cells, or less fully differentiated cells of the lymphoid lineage. Table 79–1 lists the chapters describing each of these conditions.
I. Primary lymphocytosis A. Lymphocytic malignancies 1. Acute lymphocytic leukemia (Chap. 91) 2. Chronic lymphocytic leukemia and related disorders (Chap. 92) 3. Prolymphocytic leukemia (Chap. 92) 4. Hairy cell leukemia (Chap. 93) 5. Adult T-cell leukemia (Chaps. 92 and 104) 6. Leukemic phase of B-cell lymphomas (Chap. 95) 7. Large granular lymphocytic leukemia (Chap. 94) a. Natural killer (NK) cell leukemia (Chap. 104) b. CD8+ T-cell large granular lymphocytic leukemia c. CD4+ T-cell large granular lymphocytic leukemia d. γ/δ T-cell large granular lymphocytic leukemia B. Monoclonal B-cell lymphocytosis17 (Chap. 92) C. Persistent polyclonal B cell lymphocytosis26,29 II. Reactive lymphocytosis A. Mononucleosis syndromes (Chap. 82) 1. Epstein-Barr virus55 2. Cytomegalovirus58 3. HIV164 (Chap. 81) 4. Herpes simplex virus type II 5. Rubella virus165 6. Toxoplasma gondii117 7. Adenovirus 8. Infectious hepatitis virus166 10. Human herpes virus type 6 (HHV-6)169 11. Human herpes virus type 8 (HHV-8)170 12. Varicella zoster virus165 B. Bordetella pertussis62 C. NK cell lymphocytosis72 D. Stress lymphocytosis (acute)91 1. Cardiovascular collapse171 a. Acute cardiac failure b. Myocardial infarction 2. Staphylococcal toxic shock syndrome172 3. Drug-induced89 4. Major surgery 5. Sickle cell crisis173 6. Status epilepticus 7. Trauma E. Hypersensitivity reactions 1. Insect bite101 F. Persistent lymphocytosis (subacute or chronic) 1. Cancer112 2. Cigarette smoking51 3. Hyposplenism116 4. Chronic infection a. Leishmaniasis174 b. Leprosy c. Strongyloidiasis75 |
Although patients with lymphocytosis secondary to lymphoproliferative disease generally maintain abnormal lymphocyte counts that rise over time, this finding is not invariable. Patients with large granular lymphocytic leukemia (Chap. 94) may have only transient lymphocytosis that is induced by stress or exercise.
The advent of multiparameter flow cytometric and molecular diagnostic techniques has identified a syndrome in patients who have expanded populations of monoclonal B cells without other associated clinical signs or symptoms.3 This condition, monoclonal B-cell lymphocytosis (MBL) has generated a series of clinical and biologic studies investigating the prognosis and implications of this condition. An absolute B-cell count of less than 5.0 × 109/L rather than the absolute lymphocyte count is used to distinguish MBL from CLL (Chap. 92).4 This threshold is essentially arbitrary and is not based on objective clinical outcome data. MBL could be diagnosed in two situations: in subjects with a normal lymphocyte count via a screening assay (screening MBL) or during a clinical evaluation of lymphocytosis (clinical MBL).5,6,7,8 Screening MBL, also commonly referred to as low-count MBL (<500 monoclonal B cells per μL) is diagnosed when high-sensitivity flow cytometric techniques are used in unaffected sibling families with a genetic predisposition to CLL.9 Prevalence of screening MBL increases with age from 2.1 percent in individuals between 40 and 60 years of age up to 5 percent in individuals older than age 60 years.10 Single-cell analysis in familial CLL kindreds showed oligoclonality, suggesting a model of stepwise progression to CLL.11 MBL also has been detected in blood donors, with a prevalence of 6.0 to 8.3 percent in donors age 45 years or older.12 This study detected presence of a MBL clone in 149 of 2098 donors, showing that MBL prevalence is much higher in blood donors than previously reported.13 This finding has generated interest given a meta-analysis showing higher risk of non-Hodgkin lymphoma and CLL in patients who received blood transfusions.14 Individuals with known clinical MBL should not be considered suitable for blood donation. Whether this applies to screening MBL is a matter of investigation. The 10 percent prevalence of screening MBL among relatives of patients with CLL has led to questions concerning their suitability as stem cell donors for patients with CLL requiring allogeneic stem cell transplantation.15
Clinical MBL is more commonly encountered in clinical practice when patients are evaluated for lymphocytosis. A prospective study evaluated classic and new prognostic markers (IGHV mutational status and chromosomal abnormalities) in patients with clinical MBL and Rai stage 0 CLL.16 No significant differences were found either in IGHV/IGHD/IGHJ usage between the two patient groups. Similar gene and microRNA (miRNA) signatures were seen in both groups suggesting that the two conditions have an indistinguishable biologic profile but differ only in the initial size of the monoclonal population.17 This condition is biologically indistinguishable from CLL. Given the seriousness of a diagnosis of CLL, investigators have sought to evaluate how the B-cell clone relates to the clinical outcome of development into CLL.16,18 The consensus is that the risk of progression requiring CLL-specific treatment among individuals with clinical MBL is 1 to 2 percent per year compared to 5 to 7 percent per year for individuals with Rai stage 0 CLL.6,10,19,20,21,22 In contrast to this quantifiable progression for clinical MBL, progression to CLL is extremely rare among individuals with screening MBL.23 In addition, a cohort study showed that clinical MBL was an independent risk factor for hospitalization for infection after controlling for age and gender compared to a control cohort.24 Thus, individuals with clinical MBL should be followed with a physical examination and complete blood counts by a hematologist every 6 to 12 months, while longer followup intervals of 12 to 18 months are recommended in screening MBL. Clinical MBL should also be counseled about screening for second primary malignancies. Table 79–2 lists the features of screening MBL and clinical MBL.
| Clinical MBL | Screening MBL | |
|---|---|---|
| Risk of transformation to CLL-requiring therapy | 1–2% per year | Extremely rare |
| Hematologic followup interval | 6–12 months | 12–18 months |
| Risk of infections | Yes | No |
| Eligible for blood donation | No | Yes |
| Eligible for stem-cell donation | No | No |
Persistent polyclonal B-cell lymphocytosis (PPBL) is defined as a chronic, moderate increase in absolute lymphocyte counts (>4 × 109/L) without evidence for infection or other conditions that can increase the lymphocyte count.25 This type of lymphocytosis is a rare disorder that mostly affects middle-age women and is associated with smoking. It is characterized by the persistent expansion of CD27+immunoglobulin (Ig) M+IgD+ B cells, the presence of circulating binucleated lymphocytes and increased IgM serum levels.26,27 Such patients have an accumulation of polyclonal B cells that have an unusual binucleated appearance on the blood film.28 Specific morphologic features predictive of the diagnosis include basophilic vacuolated cytoplasm and monocytoid changes.28,29 These lymphocytes typically have low-to-negligible expression of CD5 or CD23 found in patients with CLL and are polyclonal with respect to light-chain expression and immunoglobulin heavy-chain gene rearrangements (Fig. 79–1).30,31
Figure 79–1.
Persistent polyclonal lymphocytosis of B lymphocytes. Blood film. A to C. Examples of the nuclear abnormality of lymphocytes in this disorder. The lymphocyte nucleus may be bilobed or segmented although not fully bilobed. Some are monolobed. D. Light chain analysis. Immunoenzymatic method. Cytocentrifuge cell preparation. Antikappa immunoglobulin light chain tagged with peroxidase and antilambda light chain tagged with alkaline phosphatase. Note polyclonal reactivity of lymphocytes; some cells with surface κ light chains (brownish) and some with surface λ light chains (reddish). Molecular studies did not show immunoglobulin gene rearrangement. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
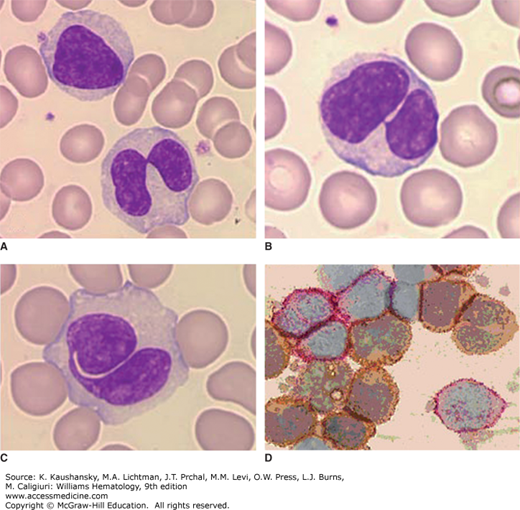
The B cells commonly express relatively high levels of IgD and CD27, a phenotype shared with that of memory B cells (Chap. 75).32 Consistent with this phenotype, the immunoglobulin variable-region genes used by the B cells most commonly have evidence of somatic mutations, implying that the expanded B cells have undergone germinal center maturation in an immune response(s) to antigen(s).33,34 Analyses of the immunoglobulin variable-region genes expressed by memory-type B cells of patients failed to reveal evidence of positive antigenic selection, suggesting that inappropriate clearance of B cells expressing low-affinity immunoglobulin receptors plays a role in this disorder.35
The cause(s) of this type of lymphocytosis is unknown. Gender and genotype may be important in the pathogenesis, as the patients most commonly are young to middle-age women who often are human leukocyte antigen (HLA)-DR7 positive.36 In addition, there are shared cases among identical twins and in families.37,38 Moreover, evaluation of first-degree relatives of individuals with this type of lymphocytosis may identify new patients who have all the criteria for its diagnosis or have slight increases in serum IgM, suggesting a possible hereditary or genetic contribution to the pathogenesis.39
Patients can have features resembling those of patients with various monoclonal B-cell malignancies. Patients may have mild splenomegaly.40 Histologic examination of marrow and secondary lymphoid tissues from patients with progressive splenomegaly can reveal features resembling marginal zone B-cell lymphoma (Chap. 101).41 In possibly another manifestation of this syndrome, first identified in Japan as hairy B-cell lymphoproliferative disorder, the patients can present with anemia, thrombocytopenia, and splenomegaly and have an excess of polyclonal B lymphocytes that appear similar in morphology and immune phenotype to the neoplastic B cells in hairy cell leukemia (Chap. 93).42,43
Although the lymphocytosis generally is not progressive, most patients have small numbers of blood B cells with chromosomal abnormalities. These abnormalities can include an additional isochromosome +i(3q) and premature chromosome condensation, and/or the t(14;18) translocation involving the BCL-2 and immunoglobulin heavy-chain loci that typically is found in the neoplastic B cells of patients with follicular lymphoma (Chap. 99).44,45,46,47 In another study of 43 patients, two-thirds of patients had lymphocytes with independent chromosomal abnormalities, such as del(6q), +der, +8, or other polyploidy karyotypic abnormalities.48,49 In any one patient, these chromosomal abnormalities are restricted to B lymphocytes independent of their expression of immunoglobulin or light chains.50 For cases associated with smoking, these cytogenetic abnormalities apparently persist after the discontinuation of tobacco use.36,51 The finding of such chromosome abnormalities is consistent with the notion that this disorder represents a preneoplastic state. Extensive proliferation of CD27+ IgM+IgD+ cells have been noted as well, which may explain the finding of splenomegaly.26 Occasional reports of clonal immunoglobulin gene rearrangements in this disorder suggest that polyclonal expansion in some cases may be followed by the emergence of one predominant clone.40 Moreover, a small proportion of patients ultimately develop monoclonal B-cell lymphoma or B-cell leukemia.40,52,53
Secondary lymphocytosis defines conditions associated with an increase in the absolute number of lymphocytes secondary to a physiologic or pathophysiologic response to infection, toxins, cytokines, or unknown factors.
The most common reactive lymphocytosis is infectious mononucleosis (see Table 79–1). In cases of mononucleosis secondary to infection with Epstein-Barr virus (EBV), the atypical lymphocytes commonly consist of polyclonal populations of CD8+ T cells, γ/δ T cells, and CD16+CD56+ NK cells that are stimulated in response to EBV-infected B cells (see Chap. 82, Fig. 82–1).54 A study prospectively evaluated university students to determine the incidence, risk factors, and virologic and immune correlates of disease severity.55 During a median of 3 years of observation of EBV antibody-negative students, 66 subjects experienced primary infection. Of these, 77 percent had infectious mononucleosis, 12 percent had atypical symptoms, and 11 percent were asymptomatic. Although viremia was transient, median oral shedding was 175 days. Increases were observed in numbers of NK cells and CD8+ T cells but not in numbers of CD4+ T cells during acute infection. Severity of illness correlated with both blood EBV load (P = 0.015) and CD8+ lymphocytosis (P = 0.0003).
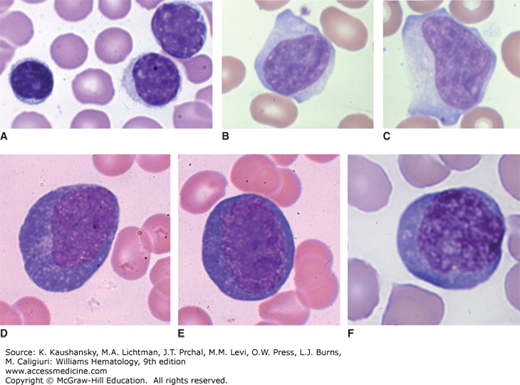
Acute infection lymphocytosis is a disorder that occurs in children usually between the ages of 2 and 10 years. It is characterized by an increase in blood lymphocytes, often to 20 to 30 × 109/L56 and occasionally as high as 100 × 109/L, which might be mistaken for acute leukemia.57 The lymphocytes may vary in size but are otherwise similar to normal blood lymphocytes (Fig. 79–2). Patients usually are asymptomatic but may have fever, abdominal pain, or diarrhea. Lymph node enlargement and splenomegaly do not occur, and the patient’s serum usually is negative for heterophile antibodies found in patients with infectious mononucleosis caused by EBV. In this regard, the disease resembles infectious mononucleosis caused by viruses other than EBV, such as cytomegalovirus (CMV; Chap. 82).58,59,60 Clinical symptoms last for a few days, but the lymphocytosis may persist for several weeks. Eosinophilia may be present. Examination of marrow from a few patients has shown minimal increases in lymphocytes, but marked infiltration with lymphocytes also has been observed. In some cases, the lymphocytosis has been found in association with acute infection by coxsackievirus B2.61
Figure 79–2.
Blood films. A. Acute infectious lymphocytosis. The lymphocytosis in this disorder of childhood is composed of normal-appearing lymphocytes, which may vary somewhat in size as shown in the blood of this case. Note typical small lymphocyte with dense chromatin pattern and scant rim of cytoplasm and somewhat two larger lymphocytes with less-dense chromatin pattern. B, C. Reactive lymphocytes. Large lymphocytes with an increased proportion of cytoplasm with basophilic cytoplasmic edges, often engaging neighboring red cells. Nucleoli may occasionally be evident. This variation in lymphocyte appearance can occur in a variety of disorders that provoke an immunologic response, including viral illnesses. They are indistinguishable in appearance by light microscopy from the reactive lymphocytes seen in infectious mononucleosis, viral hepatitis, or other conditions such as Dengue fever. D to F. Plasmacytoid lymphocytes. In this type of reactive lymphocytosis, the lymphocytes are large and have deep blue-colored cytoplasm, approaching the coloration of plasma cell cytoplasm, but they retain the nuclear appearance, cell shape, and cell size of a medium-size lymphocyte, and they do not develop a prominent paranuclear clear zone or markedly eccentric nuclear position as do most plasma cells. They may be seen in a variety of situations including infections, drug hypersensitivity, and serum-sickness-type reactions. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
A marked increase in the number of lymphocytes occurs in patients infected with the Gram-negative bacterium Bordetella pertussis.62 Absolute lymphocyte counts range from 8 to 70 × 109/L, with a mean of approximately 30 × 109/L, involving all lymphocyte subsets.63 A notable proportion of lymphocytes have cleaved nuclei, characteristic of the cells in cases of pertussis (see Chap. 73, Fig. 73–1C).
Lymphocytosis primarily results from failure of lymphocytes to leave the blood because of pertussis toxin, which is released by the bacteria.64 Pertussis toxin is an adenosine diphosphate ribosylase that modifies G proteins in mammalian lymphocytes. This inhibits the capacity of lymphocytes to traffic from blood into lymphoid tissues, primarily through inhibition of chemokine receptors. Pertussis toxin also may stimulate egress of maturing T cells from the thymus and may bind to neuraminic acid residues of T-cell surface glycoproteins to induce T-cell activation.65,66 Despite high levels of vaccination, recent epidemics have been noted primarily as a result of waning immunity in adults who subsequently serve as a source of infection to household infants.67
Large granular lymphocytosis can result from expansions of NK cells, CD8+ T cells, or, more rarely, CD4+ T cells.68,69 In the most common form, the lymphocytosis is secondary to CD3–CD16+CD56+ NK cells and is termed NK lymphocytosis, in which NK cell counts typically approximate 4 × 109/L, but can sometimes exceed 15 × 109/L.70 The blood lymphocytes of patients with T-cell large granular lymphocytosis should be evaluated for clonal rearrangements in the T-cell receptor genes (Chap. 76),51 which would be indicative of T-cell large granular lymphocytic leukemia (LGLL); LGLL is a heterogeneous disorder characterized by an increase in the number of blood large granular lymphocytes between 2 and 20 × 109/L for more than 6 months without a clearly identified cause (see Chap. 94, Fig. 94–1).71 Currently NK cell lymphoproliferative disorder is considered as a provisional entity, distinct from T-LGLL and NK-LGLL in the 2008 WHO (World Health Organization) classification. A retrospective review compared clinical and pathologic features between patients with T-LGLL and chronic NK lymphocytosis.72 They noted that median age, association with autoimmune diseases and hematologic malignancies were similar between the two groups. However, neutropenia and association with rheumatoid arthritis was less prevalent in NK cell lymphoproliferative disorder than in T-LGLL.
Large granular lymphocytosis has been observed in 20 percent of allogeneic stem cell transplant recipients for a variety of malignancies with a median onset of 312 days from transplant.73 CMV-seropositive recipients and patients who developed CMV reactivation and chronic graft-versus-host disease (GVHD) were more likely to develop large granular lymphocytosis. GVHD is a condition occurring after allogeneic hematopoietic stem cell transplantation when alloreactive T lymphocytes from the graft attack host organs, resulting in protean manifestations in multiple organs, and can cause severe debilitation. Surprisingly, presence of large granular lymphocytosis was associated with an overall survival advantage (86.2 percent vs. 53.8 percent, p<0.0001), lower nonrelapse mortality (3.2 percent vs. 27.3 percent, p<0.0001) and lower relapse incidence (9.6 percent vs. 29.4 percent, p<0.0001).
Expansion of NK cells or T cells may represent an exaggerated response to systemic infection and/or immune deregulation. T-cell large granular lymphocytosis may be secondary to an exaggerated cellular immune response to infection with human CMV.74 Also, there is an association between NK lymphocytosis and strongyloidiasis.75
Patients with NK lymphocytosis frequently have recurrent cutaneous lesions, such as livedoid vasculopathy, urticarial vasculitis, or complex recurrent aphthous stomatitis.76,77 Other reports noted an association between NK lymphocytosis and various cytopenias, including severe aplastic anemia.70,78 Large granular lymphocytosis also may be associated with rheumatoid arthritis. Occurring in less than 0.6 percent of patients with rheumatoid arthritis, large granular lymphocytic lymphocytosis almost invariably is associated with neutropenia in the absence of splenomegaly and thus may represent a subset of Felty syndrome.79,80 Patients with autoimmune pure red cell aplasia or immune thrombocytopenia also may have large granular lymphocytosis secondary to expanded numbers of polyclonal T cells or NK cells.81,82
Dasatinib and ibrutinib are associated with lymphocytosis when used for chronic myelogenous leukemia (CML) and CLL, respectively. In patients receiving dasatinib, expansion of highly differentiated CD8+ T lymphocytes or NK cells have been noted.83,84,85 Some studies associate oligoclonal expansions of these cells to clinical effects such as CMV reactivation and pleural effusion.86 Clonal lymphocytosis usually has LGLL morphology and also show late differentiated (CD27–CD57+) phenotypes that seem predisposed to apoptosis and reduced NK-cell cytotoxicity. In addition to lymphocytosis, plasma levels of interleukin (IL)-6, monokines induced by interferon-γ (IFN-γ) and IL-2R were significantly increased in LGLL patients. IFN-γ is a soluble cytokine critical for innate and adaptive immunity against viral, bacterial and protozoal infections. IL-2 is also a cytokine that has effects on T lymphocytes and has key functions in tolerance and immunity. Some studies suggest that lymphocytosis after dasatinib is associated with a favorable response in CML.87
Ibrutinib targets B-cell receptor signaling and has been approved for use in CLL.88 After just one dose of ibrutinib, increases in the absolute lymphocyte count of up to 66 percent can occur, representing egress of lymphocytes from nodal compartments.89 Although this resolves within 8 months in most patients, a small minority has sustained lymphocytosis lasting more than a year. Biologic characterization of the lymphocytosis has shown that the persistent CLL cells do not proliferate and do not represent clonal evolution. The prolonged lymphocytosis likely represents a persistent quiescent clone and is not associated with a risk of relapse.90
Transient stress lymphocytosis has been identified as a common cause of lymphocytosis in patients admitted to a hospital.91 Both trauma and nontraumatic stress have been associated with lymphocytosis.92,93 Trauma, surgery, acute cardiac failure, septic shock, myocardial infarction, sickle cell crisis, or status epilepticus may be associated with an elevated lymphocyte count, often greater than 5 × 109/L, which may revert to normal or below-normal levels within hours.94,95 The increased lymphocyte count appears promptly after the event and appears secondary to lymphocyte redistribution affecting all major lymphocyte subsets.92 A transient lymphocytosis can be induced by the redistribution of leukocyte subsets after both physical and psychological stress.96,97 Characteristically, two phases are recognized after catecholamine administration: a quick (<30 minutes) mobilization of lymphocytes, followed by an increase in granulocyte numbers with decreasing lymphocyte numbers.98,99
Delayed hypersensitivity reactions to insect bites, especially mosquitos, may be associated with a large granular lymphocytic lymphocytosis and adenopathy.100 These delayed hypersensitivity reactions can be associated with EBV-NK lymphocytosis.101 Idiosyncratic drug reactions also may be associated with subacute lymphocytosis, typically developing 2 to 8 weeks after initiating administration of the responsible drug.102,103,104,105,106,107
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


