HEREDITARY RENAL CANCER SYNDROMES
Gene for Clear Cell Kidney Cancer
Mutation of the VHL gene is found in a high percentage of tumors from patients with clear cell kidney cancer.19 Nickerson et al.20 recently identified mutation or methylation of the VHL gene in 92% of clear cell kidney cancers. VHL gene mutation is not found in papillary, chromophobe, collecting duct, medullary, or other types of kidney cancer.
Function of the VHL Protein
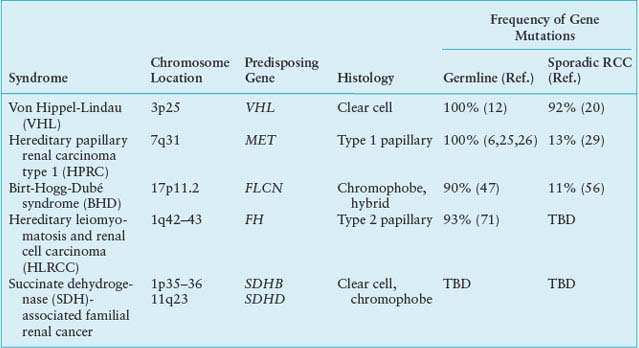
The most well-understood function of the VHL protein pVHL is the substrate recognition site for the hypoxia-inducible factor (HIF)-α family of transcription factors targeting them for ubiquitin-mediated proteasomal degradation (Fig. 20.1).14 pVHL binds through its α domain to elongin C and forms an E3 ubiquitin ligase complex with elongin B, cullin-2, and Rbx-1. Under normal oxygen conditions, HIF-α becomes hydroxylated on critical prolines by a family of HIF prolyl hydroxylases (PHD) that require 2-oxoglutarate, molecular oxygen, ascorbic acid, and iron as cofactors. pVHL then binds to hydroxylated HIF-α through its β domain, targeting HIF-α for ubiquitylation by the E3 ligase complex. Under hypoxic conditions when PHDs are unable to function or when pVHL is mutated, altering its binding to HIF-α or elongin C binding, HIF-α cannot be recognized by pVHL. HIF-α accumulates and transcriptionally up-regulates a number of genes important in blood vessel development (EPO, VEGF), cell proliferation (PDGFβ, TGFα), and glucose metabolism (GLUT-1).14 HIF-α dependent up-regulation of target genes involved in neovascularization provides an explanation for the increased vascularity of central nervous system (CNS) hemangioblastomas and clear cell renal tumors in VHL. Germline VHL mutations frequently occur in the pVHL binding domains for HIF-α and elongin C.21 HIF-2α, rather than HIF-1α, stabilization appears to be critical for renal tumor development.22,23 Additional HIF-dependent and HIF-independent functions for pVHL have been reported.10,14
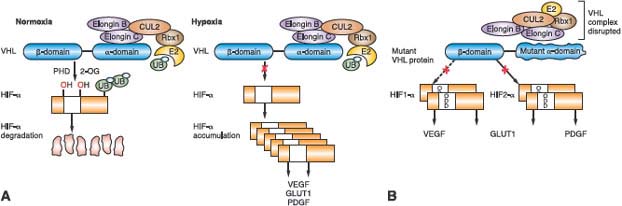
FIGURE 20.1 The von Hippel-Lindau (VHL) E3 ubiquitin ligase complex targets hypoxia-inducible factor (HIF)-α for ubiquitin-mediated degradation. A: Under normal oxygen conditions, HIF-α is hydroxylated on critical prolines by HIF prolyl hydroxylase (PHD), requiring molecular oxygen, 2-oxoglutarate, and iron as cosubstrates. The VHL protein (pVHL) can then recognize and bind hydroxylated HIF-α, enabling its ubiquitylation by the VHL E3 ligase complex and degradation by the proteasome. Under hypoxic conditions, PHD is unable to function properly, pVHL cannot recognize HIF-α, and HIF-α accumulates, leading to up-regulation of HIF-target genes (VEGF, GLUT1, PDGF) that support tumor growth and neovascularization. B: When VHL is mutated and pVHL is unable to bind HIF-α, HIF-α stabilization leads to transcriptional up-regulation of HIF target genes. (From ref. 4, with permission.)
HEREDITARY PAPILLARY RENAL CARCINOMA TYPE 1
HPRC is an autosomal dominant hereditary cancer syndrome in which affected individuals are at risk for the development of multifocal, bilateral papillary type 1 kidney cancer.24 HPRC develops in the fifth and sixth decades with age-dependent penetrance estimated at 67% by 60 years of age25; however, early onset HPRC has been described.26 This rare disorder has been reported in less than 40 kindreds worldwide.24
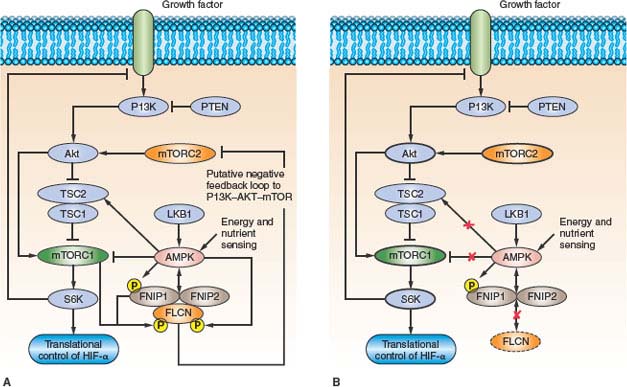
FIGURE 20.2 The putative Birt-Hogg-Dubé gene (FLCN) pathway. A: FLCN binds through FNIP1/2 to adenosine monophosphate (AMP)-activated protein kinase (AMPK) and may become phosphorylated by AMPK or by a rapamycin-sensitive kinase (i.e., mammalian target of rapamycin [mTOR]). B: When FLCN is inactivated and, presumably, FLCN protein is absent, mTOR is dysregulated, potentially driving kidney tumor formation in BHD patients. (From ref. 4, with permission.)
Genetics of Hereditary Papillary Renal Carcinoma: MET Protooncogene
In 1995 Zbar et al.27 described ten families in which multifocal, bilateral papillary renal tumors were inherited in an autosomal dominant fashion and suggested that these families might represent a hereditary counterpart to sporadic papillary tumors. Schmidt et al.6 localized the HPRC disease locus to chromosome 7q31.1-34 by linkage analysis. Since trisomy of chromosome 7 was described as a hallmark feature of papillary renal tumors,28 a gain-of-function oncogene seemed a likely candidate disease gene; in fact, germline missense mutations were identified in the tyrosine kinase domain of the MET protooncogene located at 7q31 in affected HPRC family members.6 Mutations of the MET gene have been detected in 13% of sporadic papillary renal tumors.6,29 Further studies to determine the role of MET and related genes in papillary type 1 kidney cancer are currently under way.
Hereditary Papillary Renal Carcinoma: Functional Consequences of MET Mutations
The MET protooncogene encodes the hepatocyte growth factor/scatter factor (HGF/SF) receptor tyrosine kinase. Binding of ligand HGF to MET triggers autophosphorylation of critical tyrosines in the intracellular tyrosine kinase domain, subsequent phosphorylation of tyrosines in the multifunctional docking site, and recruitment of a variety of transducers of downstream signaling cascades that regulate cellular programs, leading to cell growth, branching morphogenesis, differentiation, and “invasive growth.”30 Although MET overexpression has been demonstrated in a number of epithelial cancers,31 HPRC was the first cancer syndrome for which germline MET mutations were identified. The missense MET mutations in HPRC are constitutively activating without ligand stimulation, display oncogenic potential in vitro,32,33 and are predicted by molecular modeling to stabilize active MET kinase.34 Nonrandom duplication of the chromosome 7 bearing the mutant MET allele was demonstrated in papillary renal tumors from HPRC patients35 and may represent the second step in HPRC tumor pathogenesis. The presence of two copies of mutant MET may give kidney cells a proliferative growth advantage and lead to tumor progression.
XP11.2 TRANSLOCATION RENAL CELL CANCER
Xp11.2 translocation renal cell carcinomas, typically presenting with papillary architecture and clear or eosinophilic cytoplasm, are uncommon tumors often detected in young children and adolescents. Translocations involving Xp11.2 and 1q 21.2 associated with sporadic papillary renal carcinoma, and first described in a 2-year-old child,36 generate a fusion between a novel gene, PRCC, and the basic helix-loop-helix family transcription factor gene, TFE3.37 The encoded fusion protein, PRCC-TFE3, acts as a stronger transcriptional activator than native TFE3, and loss of the majority of native TFE3 transcripts is observed in these tumors. This deregulation of normal TFE3 transcriptional control caused by the chromosomal translocation may be important to the development of sporadic papillary renal cell carcinoma.38,39 Xp11.2 translocation renal cell carcinomas involving at least five different TFE3 gene fusions and resulting in deregulation of TFE3 transcription activity have been described, including the identical ASPL-TFE3 fusion associated with alveolar soft part sarcoma.40 Tsuda et al.41 have shown that these TFE3 fusion proteins are strong transcriptional activators of the MET gene, resulting in inappropriate MET-directed cell proliferation and invasive growth. Given the physiologic consequences of TFE3 fusion protein expression, therapeutic targeting of MET may be an effective treatment for Xp11.2 translocation renal tumors.
BIRT-HOGG-DUBÉ SYNDROME
BHD syndrome is a rare autosomal-dominant inherited cancer syndrome characterized by benign tumors of the hair follicle (fibrofolliculoma), pulmonary cysts and spontaneous pneumothorax, and a sevenfold increased risk for renal cancers.42–45 Fibrofolliculomas and lung cysts are the most common manifestations (>85% of BHD patients).46–48 Renal tumors with variable histologies develop in about 30% of BHD-affected individuals (median age 48 to 50 years), most frequently chromophobe renal carcinoma and hybrid oncocytic tumors.46,49 Metastases may develop from BHD renal tumors, but they are uncommon.
Genetics of Birt-Hogg-Dubé: Folliculin Gene
Linkage analysis performed in BHD kindreds led to localization of the disease locus to the short arm of chromosome 1750,51 and the identification of the BHD gene, FLCN.8 Almost all BHD-associated FLCN mutations are predicted to truncate the BHD protein, folliculin, including insertion or deletion, nonsense, and splice-site mutations,8,46,47,52 but recently several missense mutations located in conserved amino acid residues have been described.47,53 The mutation detection rate in several large BHD cohorts approached 90%, and germline mutations were distributed throughout the entire length of the FLCN gene with no clear genotype-phenotype correlations.46,47,54 Vocke et al.55 identified second “hit” somatic mutations or LOH in 70% of renal tumors from BHD patients, supporting a role for FLCN as a tumor suppressor gene that predisposes to renal tumors when both copies are inactivated. Gad et al.56 detected FLCN mutations in 11% of chromophobe renal cell carcinomas; others found infrequent FLCN mutations in other histologic variants of RCC.57–59 Further studies are currently in progress to evaluate the role of FLCN and related genes in chromophobe kidney cancer.
Function of the Birt-Hogg-Dubé Protein, Folliculin
The function of the BHD protein, FLCN, is currently under investigation. Baba et al.60 identified a novel folliculin interacting protein, FNIP1, and showed that FNIP1 interacts with the γ subunit of 5’ adenosine monophosphate (AMP)-activated protein kinase (AMPK), an energy sensor in cells that negatively regulates mammalian target of rapamycin (mTOR), the master switch for protein translation and cell proliferation, through TSC1/2.61,62 A second folliculin interacting protein, FNIP2, was subsequently identified that displayed similar biochemical properties to FNIP1.63,64 FLCN, through FNIP1/2, may play a role in regulation of the AMPK-TSC1/2-mTOR signaling pathway (Fig. 20.2). Published data supporting mTOR activation65,66 as well as mTOR inhibition67–69 as a consequence of FLCN inactivation in in vivo models and BHD renal tumors has led to the hypothesis that the mechanism by which FLCN interacts with and modulates mTOR is context dependent.68
HEREDITARY LEIOMYOMATOSIS AND RENAL CELL CARCINOMA
HLRCC is an autosomal dominantly inherited disorder that predisposes to the development of skin and uterine leiomyomas and an aggressive type 2 papillary renal carcinoma. Fewer than 150 HLRCC families have been reported worldwide.70,71 Renal tumors, which are often unilateral and solitary,70,72 may develop with early age of onset in 15% to 62% of affected individuals70,71 and can be aggressive, metastasize, and cause death within 5 years of diagnosis.
Genetics of Hereditary Leiomyomatosis and Renal Cell Carcinoma: Fumarate Hydratase Gene
Linkage localized the HLRCC disease locus to chromosome 1q42-43,73 but an association with renal cancer was not appreciated until Launonen et al.74 demonstrated linkage to chromosome 1q in two Finnish MCUL kindreds with solitary, highly aggressive papillary type 2 renal tumors. The disorder was renamed hereditary leiomyomatosis and renal cell carcinoma and the locus was subsequently mapped to a 1.6Mb region of 1q42. Germline mutations were identified in the fumarate hydratase (FH) gene, a Krebs cycle enzyme that converts fumarate to malate in HLRCC-affected family members.7 FH mutations in HLRCC include missense, frameshift, nonsense, and splice-site mutations as well as partial and complete gene deletions.70,72,75,76 Missense mutations are most common (57%) and occur mainly at evolutionarily conserved residues.72,75,76 Mutations are found throughout the entire length of the FH gene excluding exon 1, which encodes a mitochondrial signal peptide, and no clear genotype–phenotype associations have been reported.70 FH acts as a classic tumor suppressor gene with loss or somatic mutation of the wild type FH allele at high frequency in renal tumors and skin and uterine leiomyomata.7 FH mutations are rarely detected in sporadic uterine and skin leiomyomata or sporadic RCC.77
Functional Consequences of Fumarate Hydratase Mutations
FH mutations reduce FH activity by 20% to 80%7,75,78 in lymphoblastoid cell lines from HLRCC patients. HLRCC-associated missense mutations significantly lowered FH activity compared with truncating mutations,75 suggesting that mutant FH monomers might act in a dominant negative manner to alter proper conformation of FH tetramers. Loss of FH activity in HLRCC leads to accumulation of fumarate and, to a lesser extent, succinate, due to a block in the Krebs cycle.79,80 Pollard et al.79 have confirmed that the accumulation of fumarate and succinate resulted in elevation of HIF-1α and HIF-target genes (VEGF, BNIP), and increased microvessel density81 in HLRCC-associated uterine fibroids. Isaacs et al.80 showed that stabilization of HIF-1α resulted from competitive inhibition of the HIF PHD cosubstrate, 2-oxoglutarate, by fumarate accumulation, leading to abrogation of PHD function and release of HIF-1α from proteasomal degradation. This pseudohypoxic drive, resulting from loss of FH activity, HIF-1α stabilization, and up-regulation of HIF-inducible genes explains the aggressive nature of HLRCC-associated renal tumors (Fig. 20.3). Recently Sudarshan et al.82 demonstrated that FH mutations in an HLRCC-derived cell line led to glucose-mediated generation of reactive oxygen species (ROS) and ROS-dependent HIF-1α stabilization, supporting an alternate mechanism by which pseudohypoxic drive could support renal tumorigenesis in HLRCC.
FIGURE 20.3 Fumarate hydratase (FH) and succinate dehydrogenase (SDH) deficient kidney cancer. FH and SDH subunit B and D mutations reduce FH and SDH enzyme activity, leading to the accumulation of fumarate and succinate, which inhibits hypoxia-inducible factor (HIF) prolyl hydroxylase (PHD) by competitive inhibition of 2-oxoglutarate (2-OG). Consequently, HIF-α is not hydroxylated, not recognized by pVHL, and accumulates. This pseudohypoxia is associated with the development of aggressive tumors that develop in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) and SDH-familial renal cancer. (From ref. 4, with permission.)
FAMILIAL RENAL CANCER: SUCCINATE DEHYDROGENASE GENE
Bilateral multifocal renal tumors with early onset (<40 years of age) have been reported in the setting of hereditary head and neck paragangliomas (HPGL) and adrenal or extra-adrenal pheochromocytomas.9 Most frequently, clear cell RCC develops; however, chromophobe RCC, papillary type 2 RCC, and renal oncocytoma have been described.83–85
Genetics of Familial Renal Cancer: Succinate Dehydrogenase Subunit B and D Mutations
Germline mutations in the gene encoding succinate dehydrogenase subunit D (SDHD) were initially associated with HPGL and later with familial and sporadic pheochromocytomas.86,87 Subsequently, inactivating mutations in SDHB were found in kindreds with familial pheochromocytoma only and with HPGL and one case of sporadic pheochromocytoma.88 Later early onset clear cell RCC was diagnosed in two individuals with HPGL and germline SDHB mutations.9 Renal carcinomas with various histologies were reported in patients with germline missense, frameshift, and nonsense mutations in SDHB and D.83–85,89
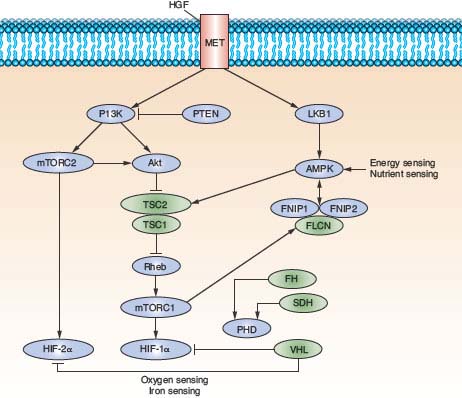
FIGURE 20.4 The genetic basis of kidney cancer. The seven renal cancer predisposing genes—VHL, MET, FLCN, FH, SDH, TSC1, and TSC2—have been identified through studies of the inherited kidney cancer syndromes. These genes interact through common nutrient and energy sensing pathways. Our understanding of the molecular mechanisms by which these genes interact in these pathways has enabled the development of targeted therapeutic agents to benefit kidney cancer patients. (From ref. 4, with permission.)
Functional Consequences of Succinate Dehydrogenase Subunit B and D Mutations
Mutational inactivation of the SDH gene results in reduced SDH enzyme activity and accumulation of succinate in renal tumors. In a mechanism similar to FH mutations in HLRCC (Fig. 20.3), accumulation of succinate serves to competitively inhibit 2-oxoglutarate and block PHD activity.79,80 In the absence of PHD, HIF-α accumulates and drives transcriptional activation of HIF-α target genes that support tumor neovascularization, growth and invasion.
TUBEROUS SCLEROSIS COMPLEX
The tuberous sclerosis complex (TSC) is a multisystem, autosomal dominant disorder affecting both children and adults and is characterized by facial angiofibromas, renal angiomyolipomas, lymphangiomyomatosis of the lung, and disabling neurologic manifestations. The disease is phenotypically heterogeneous, and many patients have only minimal symptoms of disease.90
The predominant renal manifestations in TSC are bilateral multifocal angiomyolipomas (AML), benign tumors composed of abnormal vessels, immature smooth muscle cells, and fat cells. The lifetime risk of renal cancer in TSC patients is 2% to 3%, which is similar to the general population.90 The most common histologic type of renal tumor is clear cell; however, there are rare reports of papillary RCC, chromophobe RCC, and oncocytoma in TSC patients.90 TSC is caused by mutations in one of two genes—TSC1 that encodes hamartin91 or TSC2 that encodes tuberin92—leading to loss of TSC1/2 negative regulation of mTOR signaling.61,62,93 Drug therapy targeting the mTOR pathway may be most effective for treating TSC-associated AMLs and renal tumors.94
CONCLUSION
Seven renal cancer predisposing genes—VHL, MET, FLCN, FH, SDH, TSC1, and TSC2—have been identified through studies of the inherited renal cancer syndromes, VHL, HPRC, BHD, HLRCC, SDH-related familial renal cancer and TSC (Fig. 20.4). These studies have provided valuable insight into the genetic events that lead to the development of renal tumors and the biochemical mechanisms that contribute to their progression and, ultimately, in some cases, metastasis. These findings have enabled the development of diagnostic genetic testing and provided the foundation for the development of targeted therapeutic agents for patients with the common form of sporadic kidney cancer.
Selected References
The full list of references for this chapter appears in the online version.
4. Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol 2010;7:277.
5. Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993;260:1317.
6. Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET protooncogene in papillary renal carcinomas. Nat Genet 1997;16:68.
7. Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002;30:406.
8. Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2002;2:157.
9. Vanharanta S, Buchta M, McWhinney SR, et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 2004;74:153.
10. Nordstrom-O’Brien M, van der Luijt RB, van Rooijen E, et al. Genetic analysis of von Hippel-Lindau disease. Hum Mutat 2010;31(5):521.
14. Kaelin WG Jr. The von Hippel-Lindau tumor suppressor protein: O2 sensing and cancer. Nat Rev Cancer 2008;8:865.
15. Cohen AJ, Li FP, Berg S, et al. Hereditary renal-cell carcinoma associated with a chromosomal translocation. N Engl J Med 1979;301:592.
21. Stebbins CE, Kaelin WG Jr, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science 1999;284:455.
22. Kondo K, Kico J, Nakamura E, Lechpammer M, Kaelin W. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002;1:237.
23. Maranchie JK, Vasselli JR, Riss J, et al. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 2002;1:247.
24. Dharmawardana PG, Giubellino A, Bottaro DP. Hereditary papillary renal carcinoma type I. Curr Mol Med 2004;4:855.
27. Zbar B, Glenn G, Lubensky I, et al. Hereditary papillary renal cell carcinoma: clinical studies in 10 families. J Urol 1995;153:907.
29. Schmidt L, Junker K, Nakaigawa N, et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene 1999;18:2343.
30. Gentile A, Trusolino L, Comoglio PM. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev 2008;27:85.
31. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003;4:915.
34. Miller M, Ginalski K, Lesyng B, et al. Structural basis of oncogenic activation caused by point mutations in the kinase domain of the MET proto-oncogene: modeling studies. Proteins 2001;44:32.
35. Zhuang Z, Park WS, Pack S, et al. Trisomy 7: harboring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nat Genet 1998;20:66.
37. Sidhar SK, Clark J, Gill S, et al. The t(X;1)(p11.2;q21.2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Hum Mol Genet 1996;5:1333.
39. Weterman MA, van Groningen JJ, den Hartog A, Geurts van Kessel A. Transformation capacities of the papillary renal cell carcinoma-associated PRCCTFE3 and TFE3PRCC fusion genes. Oncogene. 2001;20:1414.
41. Tsuda M, Davis IJ, Argani P, et al. TFE3 fusions activate MET signaling by transcriptional up-regulation, defining another class of tumors as candidates for therapeutic MET inhibition. Cancer Res 2007;67:919.
42. Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 1977;113:1674.
43. Toro JR, Glenn GM, Duray PH, et al. Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol 1999;135:1195.
44. Zbar B, Alvord WG, Glenn GM, et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé syndrome. Cancer Epidemiol Biomarkers Prev 2002;11:393.
46. Schmidt LS, Nickerson ML, Warren MB, et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dubé syndrome. Am J Hum Genet 2005;76:1023.
47. Toro JR, Wei MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 2008;45:321.
49. Pavlovich CP, Walther MM, Eyler RA, et al. Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 2002;26:1542.
50. Schmidt LS, Warren MB, Nickerson ML, et al. Birt-Hogg-Dubé syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet 2001;69:876.
55. Vocke CD, Yang Y, Pavlovich CP, et al. High frequency of somatic frameshift BHD gene mutations in Birt-Hogg-Dubé-associated renal tumors. J Natl Cancer Inst 2005; 97:931.
60. Baba M, Hong SB, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A 2006;103:15552.
61. Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 2005; 37:19.
63. Hasumi H, Baba M, Hong SB, et al. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene 2008;415:60.
65. Baba M, Furihata M, Hong SB, et al. Kidney-targeted Birt-Hogg-Dubé gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst 2008;100:140.
66. Hasumi Y, Baba M, Ajima R, et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci U S A 2009;106:18722.
67. van Slegtenhorst M, Khabibullin D, Hartman TR, et al. The Birt-Hogg-Dubé and tuberous sclerosis complex homologs have opposing roles in amino acid homeostasis in Schizosaccharomyces pombe. J Biol Chem 2007;282:24583.
68. Hudon V, Sabourin S, Dydensborg AB, et al. Renal tumor suppressor function of the Birt-Hogg-Dubé syndrome gene product folliculin. J Med Genet 2010;47(3):182.
69. Hartman TR, Nicolas E, Klein-Szanto A, et al. The role of the Birt-Hogg-Dubé protein in mTOR activation and renal tumorigenesis. Oncogene 2009;28:1594.
70. Kiuru M, Launonen V. Hereditary leiomyomatosis and renal cell cancer (HLRCC). Curr Mol Med 2004;4:869.
71. Wei MH, Toure O, Glenn GM, et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 2006;43:18.
73. Alam NA, Bevan S, Churchman M, et al. Localization of a gene (MCUL1) for multiple cutaneous leiomyomata and uterine fibroids to chromosome 1q42.3-q43. Am J Hum Genet 2001;68:1264.
74. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 2001;98:3387.
75. Alam NA, Rowan AJ, Wortham NC, et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet 2003;12:1241.
79. Pollard PJ, Briere JJ, Alam NA, et al Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 2005;14:2231.
80. Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 2005;8:143–53.
82. Sudarshan S, Sourbier C, Kong HS, et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol 2009;29:4080.
85. Ricketts C, Woodward ER, Killick P, et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 2008;100:1260.
90. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006;355:1345.
91. van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997;277:805.
92. The European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993;75:1305.
93. Shaw RJ, Bardeesy N, Manning BD, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 2004;6:91.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


