INTRODUCTION
SUMMARY
The innate immune system provides immediate protection against infection and serves an essential antigen-presenting role that allows the adaptive immune response to occur during the days and weeks that follow. The sensory apparatus that allows detection of infectious microbes has been deciphered in large part, and it is now known that Toll-like receptors, NOD-like receptors, RIG-I–like helicases, C-type lectin receptors, and cytosolic sensors of DNA, most notably cyclic guanosine monophosphate/adenosine monophosphate synthetase, permit recognition of specific molecules of microbial origin. Much has also been learned of the biochemical events that follow activation of these sensors. Susceptibility to infection in humans is strongly heritable, and among the many loci that influence it, those that encode proteins vital to the innate immune response are of central importance. Moreover, autoinflammatory and autoimmune diseases are dependent upon the activation of innate immune signaling pathways.
Acronyms and Abbreviations
BIR, baculovirus inhibitor of apoptosis repeat; CARD, caspase activating and recruitment domain; CD, cluster of differentiation; cGAS, cyclic AMP/GMP synthetase; CTLA, cytotoxic T-lymphocyte antigen; DAI, DNA-dependent activator of IRFs; ERK, extracellular signal-regulated kinase; FADD, Fas-associated death domain; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-monocyte colony-stimulating factor; IFN, interferon; IκB; inhibitor of κB; IKK, IκB kinase; IL, interleukin; IPAF, ice-protease activating factor; IPS-1, IFN-β promoter stimulator 1; IRAK, interleukin-1 receptor-associated kinase; IRF, interferon response factor; JAK, Janus kinase; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharide; LRR, leucine-rich repeat; MAL, MyD88 adaptor-like; MDA5, melanoma differentiation-associated gene 5; MDP, muramyl dipeptide; MyD88, myeloid differentiation primary response 88; NACHT, a nucleotide-binding domain present in NAIP, CIITA, HET-E, and TP-1; NADPH, nicotinamide adenine dinucleotide phosphate; NBS, nucleotide binding sequence; NEMO, NF-κB essential modulator; NF-κB, nuclear factor-κB; NK, natural killer; NLR, NOD-like receptor; NOD, nucleotide-binding oligomerization domain; PAR-2, proteinase-activated G-protein–coupled receptor; PRAT4A, protein associated with TLR4; PYD, pyrin domain; RIG-I, retinoic acid inducible gene I; RIP, receptor-interacting protein; RLH, RIG-I–like helicase; ROS, reactive oxygen species; SARM, sterile-α and armadillo motif; SOCS-1, suppressor of cytokine signaling 1; STAT, signal transducer and activator of transcription; STING, stimulator of interferon genes; TAK-1, transforming growth factor-β–activating kinase 1; TBK1, TANK-binding kinase 1; TIR, Toll/interleukin-1 receptor; TLR, Toll-like receptor; TNF, tumor necrosis factor; Tpl2, tumor progression locus 2; TRAF, TNF receptor–associated factor; TRAM, TRIF-related adaptor molecule; TRIF, Toll/interleukin-1 receptor (TIR) domain-containing adaptor inducing IFN-β; UCM, upregulation of costimulatory molecules.
INNATE IMMUNITY VERSUS ADAPTIVE IMMUNITY
In humans, as in all mammals, resistance to microbial infection is based partly upon lymphocytes, which yield highly specific responses to microbial antigens: either the production of antibodies or the expansion of T-cell cell clones that are directly cytotoxic to infected cells (Chaps. 75 and 76). This, the adaptive immune response, is a recent fixture in evolution, witnessed only in vertebrates and traceable to the development of a mechanism for recombination of genomic DNA that arose approximately 450 to 500 million years ago, operating on genes encoding proteins with immunoglobulin domains in some lineages and on genes encoding proteins with leucine-rich repeats in other lineages.1,2 A more fundamental type of immunity, known as innate immunity, is represented in one form or another in all multicellular organisms. For this reason, a great deal of progress in the innate immunity field has come from the study of model animals such as Drosophila melanogaster, and model plants such as Arabidopsis thaliana. Despite the vast evolutionary divergence of these organisms from Homo sapiens, these species use defensive proteins and signaling pathways that are ancestrally related to those represented in humans.
Like the adaptive immune system, the innate immune system is endowed with a means of detecting microbes, destroying them, and at the same time, exercising self-tolerance. These mechanisms are far older than the analogous adaptive mechanisms and as a consequence are more refined. Although it is sometimes termed the “primitive” immune system, the innate immune system is both sophisticated and highly effective. Moreover, adaptive immunity is largely dependent upon innate immunity in the sense that antigen presentation and adaptive immune activation depend upon innate immune cells.
Innate immunity, which acts immediately to protect the host in the event of microbial inoculation, fills a temporal gap that would otherwise exist in the global immune response. Days or weeks are required for an effective adaptive immune response to develop when the naïve host encounters a new pathogen. During this time, innate immunity alone protects the host. Indeed, innate immunity is objectively more important than adaptive immunity. In a nonsterile environment, survival would be impossible without it (Table 20–1).
| Innate Immunity | Adaptive Immunity | |
|---|---|---|
| Sensing mechanism | TLRs, NK receptors; NLRs, RLHs, fMLP receptor | Immunoglobulins, T-cell receptors |
| Cellular components | Macrophages, dendritic cells, granulocytes, mast cells, NK cells | T cells, B cells |
| Efferent mechanisms | Cytokine production, inflammatory response, phagocytosis, pathogen killing | Antibody production, cytokine production, cell killing |
| Purpose | Alert other innate and adaptive immune cells to pathogen presence; directly kill pathogen; encourage the development of an adaptive immune response | Assist in efficacy of innate immune response, produce highly specific ligands for pathogens |
| Time scale of response | Quick (maximal in minutes to hours) | Slow (maximal in days to weeks) |
| Specific memory | No | Yes |
| Phylogeny | Ancient (all multicellular organisms) | Recent (vertebrates only) |
TYPES OF INNATE IMMUNITY
Innate immunity embraces a large number of host resistance mechanisms, which may be divided into cellular and noncellular components, and also into afferent and effector components. Noncellular components of innate immunity include antimicrobial peptides, which selectively disrupt microbial cell membranes, complement, components of which also disrupt cell membranes, and the proteins hemopexin and haptoglobin, which deny iron to invasive microbes. Cellular components include myeloid cells (granulocytes, monocyte/macrophages, mast cells, and dendritic cells) and lymphoid cells (natural killer [NK] cells and NKT cells). As such, it can be seen that despite their recent evolutionary origin, some lymphoid cells have been coopted to serve in the innate immune system rather than the adaptive immune system. Many other cells are also endowed with some degree of innate (often “cell-autonomous”) immune function. For example, fibroblasts can sense viral infection and respond with interferon (IFN) production.
Once initiated, the innate immune response runs its course in a preprogrammed fashion, proceeding from microbe sensing all the way through to microbial killing, making division into “afferent” and “effector” functions somewhat arbitrary. Nonetheless, the proteins responsible for microbial recognition, signaling, and the development of a transcriptional response within innate immune cells are generally considered “afferent” components; the cytokines that mediate the response and the cellular weaponry that is used to destroy viruses and bacteria may be considered “effector” components.
The remainder of this chapter emphasizes the afferent arm of cellular innate immunity, as the effector mechanisms (neutrophil-mediated killing, complement, and antimicrobial peptides) are covered in other chapters. Our understanding of innate immune responses has improved dramatically as forward and reverse genetic methods have been used to dissect the signaling pathways that permit host recognition of microbes. The initial interactions between molecules of microbes and molecules of the host that trigger an innate immune response have been studied in great detail over the past decade. The afferent pathways are each capable of activating responses that partly overlap with one another.
The Toll-like receptors (TLRs) collectively mediate the recognition of most microbes. Ten TLRs are encoded in the human genome. The molecular specificity of nine of these TLRs has been established, at least in part. Although publications can be found to suggest that some of the TLRs (notably TLRs 2 and 4) detect dozens of molecules, the evidence favoring most of these interactions is slender, and a conservative viewpoint is preferred; hence, Table 20–2 presents only those interactions that are deemed certain.
| TLR | Known Macromolecular Associations | Ligand(s) | Adapter Use | Refs. |
|---|---|---|---|---|
| 1 | TLR2 | Tri-acyl lipopeptides | MyD88, MAL | 12,137,138,139 |
| 2 | TLRs 1 or 6, or homodimer | Lipopeptides, lipoteichoic acid, zymosan, protozoal GPI | MyD88, MAL | 11 |
| 3 | – | dsRNA | TRIF | 14,42,120 |
| 4 | CD14, MD-2 | LPS | MyD88, MAL, TRIF, TRAM | 7,21,41,42,120,140 |
| 5 | – | Flagellin | MyD88 | 13 |
| 6 | TLR2 | Di-acyl lipopeptides, glucans, lipoteichoic acid | MyD88, MAL | 141 |
| 7 | – | ssRNA, imidazoquinolines | MyD88 | 142 |
| 8 | – | ssRNA, imidazoquinolines | MyD88 | 143 |
| 9 | – | Unmethylated CpG motifs | MyD88 | 10 |
| 10 | – | Unknown | Unknown | 144 |
The microbe-sensing function of the mammalian TLR was discovered as a result of inquiry into the mechanism of endotoxin sensing. Endotoxin (later identified as lipopolysaccharide [LPS]) was first described by Pfeiffer as a toxic component of Vibrio cholerae more than 100 years ago.3 Its chemical structure was established many years later (reviewed in Ref. 4), and a toxic “lipid A” moiety of LPS was synthesized artificially in 1985 and found to have full biologic activity.5 The identity of the LPS receptor was established in 1998, through the positional cloning of Lps, a locus that was known to be required for all cellular responses to endotoxin, and for the effective clearance of Gram-negative bacterial infections6 in laboratory mice. In LPS-unresponsive mice, the Tlr4 locus was shown to be mutationally altered or deleted.7 It had previously been recognized that Toll, a Drosophila protein also known for its developmental effects,8 was required for the innate immune response to fungal infection in flies.9 Hence, the discovery of an LPS-sensing function for TLR4, a homologue of Toll, made evolutionary sense.
Other molecules of microbial origin (for example, di- and tri-acylated lipopeptides and lipoproteins, lipoteichoic acid, unmethylated DNA bearing CpG dinucleotides in a particular context, flagellin, and double-stranded RNA [dsRNA]) were known to elicit responses qualitatively similar to those elicited by LPS. The other TLR paralogs seemed excellent candidate receptors for these molecules. Reverse genetic methods established that each of these molecules is indeed recognized by a particular TLR or heteromeric combination of TLRs.10,11,12,13,14 Moreover, genetic complementation analyses have shown that at least some microbial ligands directly engage the TLRs in order to elicit a signal.15,16 On the other hand, other molecules enhance the signal, and also participate in ligand recognition. Dectin-1 is a type II transmembrane C-type lectin that recognizes glucans present in the cell walls of fungi, signals via spleen tyrosine kinase (Syk) and the Card9/Bcl-10/MALT1 complex to activate nuclear factor-κB (NF-κB),17,18 and enhances TLR2/6 signaling.19 Similarly, proteinase-activated G-protein–coupled receptor (PAR-2) signaling enhances TLR4 responses to LPS.20 Other examples include the binding of cluster of differentiation (CD) 14 to LPS21 which augments LPS responses,22 as well as the enhancement of responses to bacterial diacylglycerides by CD36.23 It is likely that these accessory molecules form complexes with the TLRs, which are responsible for transducing the signal across the cell membrane. TLR4 exists in a tight complex with MD-2, a small secreted protein that is required for TLR4 to reach the cell surface and required for LPS sensing as well.24
The TLRs are single-spanning transmembrane proteins with leucine-rich repeat (LRR) motifs in their extracellular domains and a characteristic TIR (Toll/interleukin [IL]-1 receptor) motif in their cytoplasmic domains. The TIR domain is based on an ancient protein fold25 evident in cytosolic plant disease resistance proteins (where it often is represented together with a nucleotide binding sequence [NBS] and/or LRR motifs), in proteins of the IL-1 and IL-18 receptor family, in the adapter proteins that carry signals from TLRs, and in the TLRs themselves.
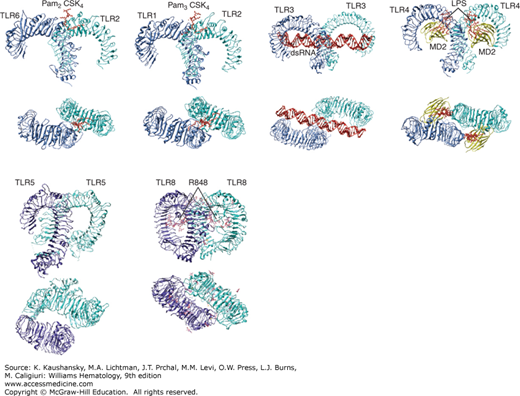
The structure of the TLR2/1, 2/6, 3, 4, 5, and 8 ectodomains has been determined by x-ray crystallography, which showed a horseshoe-shape characteristic of LRR-containing proteins. TLRs form homodimers or heterodimers induced by the simultaneous binding of ligands to LRRs of distinct receptor chains. The nature of the ligand-receptor interaction has also been determined for several of the above receptors, and appears to be different in each individual case (Fig. 20–1). To activate TLR4, LPS interacts with MD-2, which has a hydrophobic pocket that accommodates the lipid A moiety of LPS.26,27 TLR2/1 heterodimers are “crosslinked” by the engagement of two acyl chains by TLR1 and a single acyl chain by TLR2.28 TLR3 molecules bind a linear, negatively charged dsRNA oligonucleotide, which triggers activation.29
Figure 20–1.
Structures of Toll-like receptors (TLRs) and their ligands. TLR2-TLR6-Pam2CSK4 lipopeptide (3A79), TLR2-TLR1-Pam3CSK4 lipopeptide (2Z7X), TLR3-dsRNA (3CIY), TLR4-MD2-LPS (3FXI), TLR5 (3J0A), and TLR8-R848 (3W3L) are shown. Side view (upper panels) and top view (lower panels) are shown. Protein Databank ID numbers are indicated in parentheses. Figures were generated with UCSF Chimera. dsRNA, double-stranded RNA.
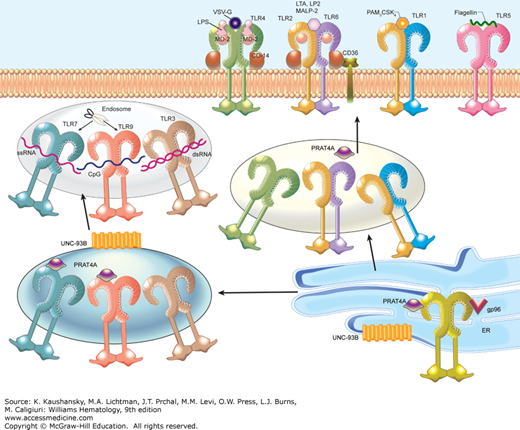
TLRs 3, 7, 8, and 9 are believed to be intracellular. Little (TLR3) or no (TLRs 7 and 9) surface expression can be detected, and tagged versions of the molecules are found to reside within the interior of transfected cells.30 The ectodomains of these TLRs project into endocytic vesicles and there detect foreign molecules rather than within the extracellular space. TLRs 3, 7, 8, and 9 are trafficked from the endoplasmic reticulum (ER) to endosomal compartments via the secretory pathway, and depend on the aid of chaperones to do so. For example, UNC93B1, a 12-transmembrane spanning ER protein, directly binds and is necessary for TLRs 3, 7, and 9 to gain access to the endosomal compartment.31 UNC93B1 is believed to escort these molecules, and perhaps others, to their destination in the cell.32 PRAT4A (encoded by TNRC5) serves a critical role in chaperoning multiple TLRs to their destination,33 while the ER chaperone protein, gp96 (also called GRP94 or HSP90B1) is critical for all TLR maturation (Fig. 20–2).34 Proteolysis of TLR7 and 9 is known to occur in the endolysosome, and at least for TLR9, this cleavage increases ligand binding and is necessary for activating downstream signaling pathways.35,36 In plasmacytoid dendritic cells, TLR7 and TLR9 are further trafficked from endosomes to lysosome-related organelles; this trafficking is necessary for the abundant production of type I IFN for which these cells are specialized.37,38 The adaptor protein complex 3 (AP-3), which directs subcellular trafficking through the secretory pathway, and the peptide/histidine transporter 1 (PHT1) are necessary for TLR7 and TLR9 trafficking to lysosome-related organelles.
Figure 20–2.
The Toll-like receptors (TLRs). The TLRs exist in homo- or heterodimeric form and are capable of sensing diverse molecules derived from pathogenic organisms. TLRs 1, 2, 4, 5, and 6 are located at the cell surface, while TLRs 3, 7, and 9 are located in the endosome. All TLR maturation is dependent on the chaperone protein gp96 in the endoplasmic reticulum (ER). Two other ER proteins, PRAT4A and UNC93B1, play important roles in TLR trafficking. PRAT4A is necessary for TLRs 1, 2, 4, 7, and 9 responses, while UNC93B1 is required for TLRs 3, 7, and 9 trafficking. At the cell surface, a TLR4 complex composed of TLR4, MD2, and CD14 specifically binds to lipopolysaccharide (LPS) and vesicular stomatitis virus glycoprotein G (VSV-G). The TLR2/6 heterodimer, along with CD36 and CD14, recognizes diacylated lipopeptides and lipoteichoic acid (LTA). The TLR1/2 heterodimer senses triacylated lipopeptides (PAM3CSK4), and TLR5 recognizes flagellin. TLR7 is able to bind to single-stranded RNA, TLR9 to CpG DNA, and TLR3 to double-stranded RNA. Proteolysis of both TLR7 and TLR9 by lysosomal cysteine proteases including cathepsins and asparagine endopeptidase occurs in the endolysosome, and at least in the case of TLR9, is required for function. Abbreviations are as used in the text.
The signaling events initiated by the TLRs are increasingly complex and have been studied in great detail [reviewed in Refs. 39 and 40]. Figure 20-3 illustrates the pathways as they are presently understood. It must be recognized that not all TLRs operate within the same cells, nor are all cells equivalent in their responses to TLR ligation. Notably, macrophages and conventional (myeloid) dendritic cells respond to different stimuli than do lymphoid cells, or plasmacytoid dendritic cells (which are specialized for type I IFN production). Moreover, some cells not usually regarded as “professional” components of the innate immune system are capable of responding to TLR ligands in one way or another.
Figure 20–3.
Overview of Toll-like receptor (TLR) signaling pathways. Shown are the activating events downstream of TLR activation that ultimately lead to the induction of thousands of genes including those encoding tumor necrosis factor (TNF) and type I interferon (IFN), which are critical in activating innate and adaptive immune responses. TLR4 activation is shown as a prototypical model. Once TLR complexes recognize a specific molecule, they recruit combinations of adaptor proteins (MyD88 [myeloid differentiation primary response 88], TRIF [Toll/interleukin-1 receptor domain-containing adaptor inducing IFN-β], TRAM [TRIF-related adaptor molecule], MAL [MyD88 adaptor-like]) and initiate activation of downstream signaling molecules. See text for details of MyD88-dependent and TRIF-dependent signaling. When bound to vesicular stomatitis virus glycoprotein G (VSV-G), TLR4 can signal through TRAM to induce interferon response factor (IRF) 7 activation, a process that is partially dependent on TRIF (not shown). K63 and K48 ubiquitination are represented by chained circles. Proteins that are degraded are shown with a dotted outline. LP2, lipopeptide 2; LTA, lipoteichoic acid. PAM3CSK4 is a triacyl lipopeptide. Phosphorylation events are represented by P-labeled circles. Abbreviations are as used in the text.
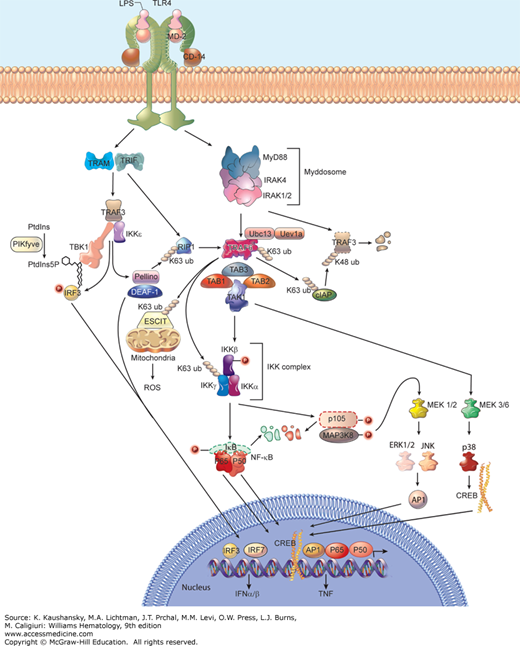
A total of five TIR adapter proteins are encoded in the human genome. These adapters are MyD88 (myeloid differentiation primary response 88), MAL (MyD88 adaptor-like; also known as TIRAP), TRIF (Toll/interleukin-1 receptor domain-containing adaptor inducing IFN-β; also known as TICAM1 and first identified by a mutant allele known as Lps2), TRAM (TRIF-related adaptor molecule; also known as TICAM2), and SARM (sterile-α and armadillo motif). The function of SARM remains unknown, and it is the most distantly related paralog among the adaptors. However, the four remaining adapters have well-defined roles in signal transduction. All four of these adapters are required for normal signaling from the LPS receptor, TLR4; MyD88 and MAL act in concert with one another, and TRIF and TRAM act together, so that two primary branches of the LPS signaling pathway diverge at the level of the receptor.41,42 In contrast, TRIF alone serves TLR3 signaling; MyD88 and MAL (but neither TRIF nor TRAM) serve TLR2; and MyD88 alone serves TLRs 7, 8, and 9. Mutational inactivation of MyD88 creates a severe immunodeficiency state in mice and humans,43,44 and compound homozygosity for mutations affecting both MyD88 and TRIF causes immunodeficiency that is still more severe, in which animals are essentially unable to sense the presence of most microbes.42
Two main branches of signaling, dependent on MyD88 or TRIF, mediate the effects of TLR activation in conventional dendritic cells, macrophages, and fibroblasts (see Fig. 20–3). The MyD88-dependent pathway is used by all TLRs except TLR3, as mentioned above. MyD88 is believed to assemble into a helical complex called the Myddosome upon receptor activation, engaging the serine kinases IRAK (interleukin-1 receptor-associated kinase) 4 and IRAK2 or IRAK1 through death domain interactions.45 Signaling proceeds via phosphorylation of IRAK2 or IRAK1 by IRAK4. No comparable structural data illuminate the function of MAL, TRIF, or TRAM proteins, but it is clear that TRIF can directly engage TLR3.46 The activated Myddosome recruits the E3 ubiquitin ligase tumor necrosis factor (TNF) receptor-associated factor (TRAF) 6, a cellular scaffold protein that coordinates the recruitment of several other protein kinases. MyD88 also interacts with TRAF3; however, degradative K48-linked ubiquitination of TRAF3 by cIAP1/2 during MyD88-dependent TLR signaling is necessary for the activation of mitogen-activated protein kinases (MAPKs) and production of inflammatory cytokines.47 In conjunction with the E2 ubiquitin-conjugating enzyme 13 (Ubc13) and the Ubc-like protein Uev1a, TRAF6 adds chains of K63-linked polyubiquitin to itself, as well as inhibitor of κB (IκB) kinase γ (IKKγ; also called NEMO [NF-κB essential modulator]) and to TRAF2 (reviewed in Ref. 48). Transforming growth factor-β–activating kinase 1 (TAK-1) forms a complex with TAB1, TAB2, and TAB3, is recruited to the TRAF6 complex, and phosphorylates IKKβ, which in complex with IKKα and IKKγ phosphorylates IκB (an inhibitor of the p65 form of NF-κB), leading to its K48-ubiquitin–mediated degradation.48 Nuclear translocation of homo- or heterodimers composed of p65 and/or p50 NF-κB ensues. NF-κB drives the transcription of hundreds of genes encoding proteins that form the inflammatory response. Mitochondrial reactive oxygen species (ROS) are also produced in macrophages as a result of TLR4, TLR2, and TLR1 activation; this antibacterial response depends on the translocation of TRAF6 to mitochondria to engage and ubiquitinate a protein called ECSIT, which functions in mitochondrial respiratory chain assembly.49
At the same time, the IKK complex activated by TAK-1 phosphorylates the p105 form of NF-κB and MAP3K8 (also known as Tpl2), proteins that form a complex in which MAP3K8 is inactive under basal conditions. This leads to the degradation of p105 NF-κB, and to the activation of MAP3K8.50,51 MAP3K8 phosphorylates and activates MEK1 and MEK2, while independently MEK3 and MEK6 are activated by TAK-1.40 The MEKs activate MAPK family members, including extracellular signal-regulated kinase (ERK) 1 and ERK2, c-Jun N-terminal kinase (JNK), and p38 kinases. These kinases trigger the activation of other transcription factors, including c-Jun, which together with c-Fos forms the transcription factor AP1, and members of the cyclic adenosine monophosphate (AMP) response element-binding protein (CREB) family.
The TRIF-dependent TLR signaling pathway is activated by TLR3 and TLR4, and results in the induction of type I IFNs as well as inflammatory response genes (see Fig. 20–3). Upon receptor activation, TRIF interacts with TRAF3, which recruits TANK-binding kinase 1 (TBK1) and IKKε (both distantly homologous to the IKKs).52,53 This complex engages and phosphorylates interferon response factor (IRF) 3, an interaction that may be mediated by phosphatidylinositol-5-phosphate generated by PIKfyve.54 IRF3 dimerizes and translocates to the nucleus to activate transcription of type I IFN genes with the aid of deformed epidermal autoregulatory factor-1 (DEAF-1).55 Two other IRF proteins, IRF1 and IRF7, also activate type I IFN genes, but in response to signaling from TLR7 and TLR9 particularly in plasmacytoid dendritic cells.56,57 Activation of IRF3 and IRF1 can initiate expression of the IFN-β gene.58,59 IFN-β mediates antiviral effects, and is also required for the upregulation of costimulatory proteins (e.g., CD40, CD80, and CD86) that enhance the activation of an adaptive immune response. Hence, the adjuvant effects of LPS and dsRNA are dependent upon the type I IFN receptor.60 IRF7 induces the expression of the IFNα genes.59,61 Both α and β IFNs bind to the type I IFN receptor rendering similar if not identical biological responses.
To induce inflammatory response genes, TRIF recruits receptor-interacting protein (RIP) 1 following its polyubiquitination by the E3 ligase Pellino.62 RIP1 interacts with the TRAF6/TAK-1 complex leading to NF-κB activation following the pathway described above for MyD88-dependent signaling. For reasons that remain unclear, the heteromeric MyD88/MAL complex is incapable of driving type I IFN gene expression.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


