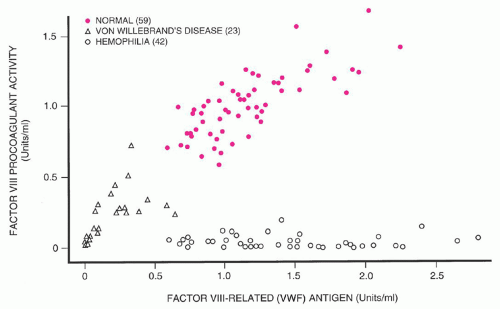
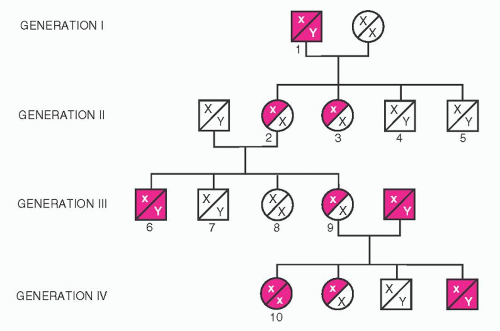
The historical confusion that has surrounded the pathogenesis of vWD is apparent from the many names that have been applied to this disorder. These designations include
angiohemophilia, vascular hemophilia, pseudohemophilia, constitutional thrombopathy, and
idiopathic prolonged bleeding time. von Willebrand first recognized the disorder in a 1926 study of the inhabitants of the Åland Islands.
125 Three cardinal manifestations of this bleeding disorder are mucocutaneous hemorrhage rather than hemarthrosis and deep-muscle bleeds; autosomal dominant inheritance, rather than sex-linked as seen in hemophilia A; and prolonged bleeding time. Evaluation of one large kindred revealed variable severity of bleeding symptoms, with some obligate heterozygote carriers being asymptomatic. von Willebrand thought that the hemostatic defect resulted from combined defects in platelet function and vascular endothelium. The discovery that plasma from normal volunteers or patients with hemophilia A corrected the hemostatic defect of vWD suggested that a plasma protein distinct from factor VIII caused vWD. In 1972, von Willebrand factor (vWF) was purified. Genetic variants were correlated with various vWF structural abnormalities, and demonstration of the heterogeneous nature of vWD was furthered by development of vWF multimer analysis by gel electrophoresis.
126 In its most common form (type 1), the disorder is characterized by mild mucocutaneous hemorrhage, which is attributable to deficiency of both vWF and factor VIIIc. The disorder is not homogeneous, however, in part because of the multiple physiologic functions played by vWF. The recognition of a number of variants, together with the demonstration of multiple genetic patterns, suggested that vWD is very heterogeneous. However, all forms of vWD can be traced to insufficient amounts of vWF or defective function of this protein. Recent reviews of this area have been published,
127,
128 and a consensus document on vWD sponsored by the National Institutes of Health reviewed all aspects of the disorder.
129,
130
Genetics
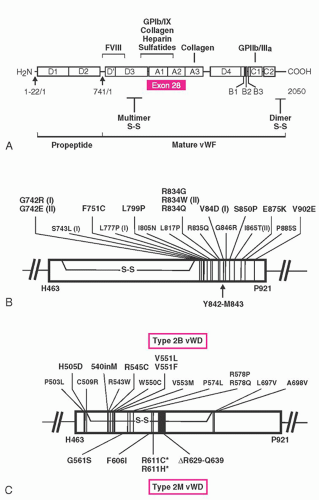
A gene on chromosome 12 codes for the synthesis of the vWF macromolecule. The genetic message is composed of 52 exons covering a span of 158 kb.
132,
133 The vWF gene encodes for a protein with multiple copies of homologous motifs, including three “A,” three “B,” two “C,” and four “D” motifs (see
Fig. 53.5). These motifs in turn encode protein domains that subserve the various functions of vWF. The A1 domain contains binding sites for platelet GPIb, ristocetin, and collagen,
134,
135 the A2 domain contains a protease-sensitive domain that may have a role in regulating vWF function,
136 the A3 domain contains a second collagen-binding domain,
135 the C1 domain has an RGD sequence capable of interacting with platelet glycoprotein IIb/IIIa, and the D’ and D3 domains contain a factor VIII-binding sequence.
135vWD appears to be inherited by multiple genetic mechanisms (
Table 53.7). The most common form of the disorder (type 1 vWD) accounts for ˜70% of all cases of vWD. Diagnostic criteria for type 1 vWD have been proposed,
128 including a history of bleeding symptoms, a positive family history of bleeding symptoms, and bleeding that is attributable to quantitatively low vWF levels. Population screening suggests that the prevalence of vWD may be as high as 1% of the population, but only a minority of these patients subsequently present with clinically significant bleeding.
137 vWD is inherited as an incompletely dominant autosomal trait with variable penetrance, even among members within a single kindred. Because of variable penetrance and expression of vWD, only ˜33% of children may be affected.
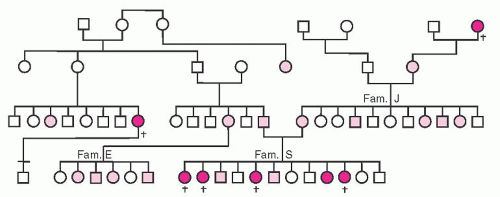
Figure 53.6 illustrates one of the original pedigrees described by von Willebrand.
Twin studies demonstrated that ˜60% of the variation in vWF level and ˜50% of the variation in factor VIII level is attributable to genetic factors.
138 Genetic loci outside of the vWF gene contribute to variation of vWF level. Individuals with blood group O have vWF levels that are on average 30% lower than those of people with blood group A, B, or AB,
139 and it is likely that carbohydrate groups attached to vWF play a subtle role in clearance of vWF from plasma.
140,
141 In addition, variation in expression of levels of other hemostatically active proteins, such as variation in the level of platelet adhesion receptors, may modulate the severity of symptoms conferred by vWF deficiency.
142 vWF levels are under hormonal and other controls, which further complicates disease expression. Variation in levels of as much as 20% has been reported with menstrual cycle, with levels lowest in the early follicular phase (before day 7 of the cycle); and levels increase with age, rising ˜15% for each decade increase in age.
143 As will be discussed later, the criteria for diagnosing quantitative vWD deficiency, and the specificity of historic bleeding, have introduced controversy in what constitutes type 1 vWD and have led to the potential for the new diagnostic entity of “low vWF”.
137,
144Mild to moderate quantitative deficiency of vWF characterizes patients with type 1 vWD, whereas virtual absence of vWF characterizes type 3 disease. Two large studies of families with a diagnosis of type 1 vWD and genetic evaluation of patients with type 3 disease have begun to shed light on the genetics of quantitative vWF deficiency.
Very low vWF levels in the range of 5 to 20 IU/dl tend to be highly inheritable and are frequently associated with “dominant negative”-acting mutations. Mutations in these patients with type 1 vWD appear to be different than those with type 3 disease, such that type 1 disease is not simply explained by being heterozygous for a type 3 allele. Genetic defects associated with these more severe type 1 phenotypes encode amino acid changes that in turn lead to defects in protein expression. Mechanisms responsible for decreased plasma vWF level include reduced secretion related to impaired intracellular transport of vWF subunits, increased clearance from the circulation, or accelerated catabolism as a result of increased susceptibility to degradation by ADAMTS13. Changes that were most likely to cause a “dominant negative” mechanism
are often missense mutations that change the number of cysteine residues in the vWF protein. Although such changes probably affect vWF synthesis and trafficking in the cell, at least one common mutation (Tyr1584Cys) also results in enhanced degradation by ADAMTS13.
145,
146 One non-cysteine-related mutation is seen in vWD Vicenza. In these patients, a mutation encoding for Arg1205His results in accelerated clearance of plasma vWF, with levels of vWF in the range of 15 IU/dl. With DDAVP stimulation, timed plasma sampling revealed that the released vWF had a circulation half-life that was approximately one-sixth of normal.
147 Several other similar mutations have recently also been described.
148At higher levels of vWF, linkage of the vWF level to the vWF gene is found less often. The recent European type 1 vWF cohort study demonstrated this observation. Almost all families with vWF levels <30 IU/dl showed linkage to the vWF gene, but the proportion fell to only 51% with vWF levels >30 IU/dl.
98 These data were corroborated in the Canadian study.
146Complete absence of vWF is responsible for the most extreme form of vWD, and this is classified as type 3 vWD.
128 As one might predict, a large variety of genetic abnormalities scattered throughout the vWF gene have been reported in families with type 3 vWD. These have included large gene deletions, small gene deletions, frame-shift mutations, splice-site mutations, nonsense mutations, and point mutations.
149Type 2 vWD is characterized by production of a qualitatively defective protein, and the genetics of these variant forms is more straightforward than that of type 1 vWD. In families affected by types 2B and 2M, as well as in the majority of families with type 2A vWD, inheritance is autosomal dominant. Rare cases of type 2A disease are transmitted in an autosomal recessive fashion, as are most cases of type 2N disease. vWF gene sequencing studies combined with vWF mutation-expression analysis has revealed that single missense point mutations underlie the majority of type 2 disorders.
128 Type 3 vWD is also inherited as a recessive trait and occurs when both copies of the vWF gene are defective. In type 3 disease, gene abnormalities include large or partial gene deletions, disruption of orderly messenger RNA transcription (frame-shift mutation, splicesite, or nonsense mutation), and missense mutation. For patients with recessively inherited vWD phenotypes (type 2N, type 3, or some rare type 2A), genetic analysis may reveal either homozygous or compound heterozygous gene abnormalities. Members of the International Society of Thrombosis and Hemostasis maintain a database of vWD mutations (www.vwf.group.shef.ac.uk).
Response to Transfusion
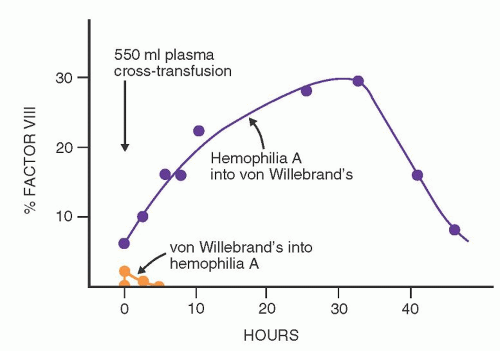
When blood products containing the vWF-factor VIII complex are infused into patients with hemophilia A, peak levels of factor VIIIc are present immediately after the infusion; this activity then declines rapidly, with an overall half-life of 8 to 10 hours (
Fig. 53.7). Twenty-four hours later, factor VIIIc activity is minimal. This response is highly predictable and is discussed in a later section concerning replacement therapy for hemophilia A. In most patients with vWD, the infusion of normal vWF-factor VIII complex produces an initial rise in factor VIIIc that is predicted from the preinfusion level and the amount of factor infused. This is followed by a sustained but variable rise in factor VIIIc activity that reaches a plateau about 24 hours later and may persist for 48 to 72 hours. This phenomenon is highly variable and irregular; in some patients, a rapid initial fall of factor VIIIc is followed by a secondary rise in activity. This disproportionate response to transfused factor VIII has been called the secondary transfusion response, a feature that is apparently unique to vWD. Various hypotheses have been advanced to explain the disproportionate response to transfusion described above. The leading hypothesis is that the infused vWF results in stimulation of increased factor VIII release into the plasma with the attachment of the newly released factor VIII onto infused vWF.
161Transfusions of blood products containing vWF shorten the bleeding time to a variable degree in patients with vWD. This corrective effect seldom persists for more than a few hours, even after massive transfusions that raise vWF to high levels. The correction of the bleeding time apparently requires the large molecular forms of vWF that are present in cryoprecipitate and select intermediate-purity factor VIII concentrates,
162 but that are completely absent from monoclonally purified or recombinant concentrates of factor VIII.
163 Failure of the skin template bleeding time of patients with vWD to show sustained correction with appropriate replacement therapy has been noted, and does not necessarily predict defective surgical hemostasis. This apparent paradox may be a result of sensitivity of the bleeding time test to platelet
α-granule vWF, which is not replenished during vWF replacement therapies.
164
Clinical Manifestations
The bleeding manifestations in vWD are heterogeneous but consistent with the dual roles of vWF in supporting both primary and secondary hemostasis. Thus, in the mild forms of vWD, the clinical picture is dominated by cutaneous and mucosal bleeding, which appears to be mainly the result of disordered primary hemostasis. In the most severe forms of the disorder, in which factor VIII levels are low, hemarthroses and dissecting intramuscular hematomas may develop. As in mild classic hemophilia, serious hemorrhage resulting from traumatic injuries or after surgical procedures is a significant hazard in severe vWD. Petechiae are rare, but hematoma formation and the extent of bruising are excessive compared with the inciting trauma.
The bleeding manifestations in the usual patient with type 1 vWD are mild, however, and many patients are virtually asymptomatic. The disorder may be symptomatic at any age, but it is not always recognized. Easy bruising is common in vWD, but it is not specific. Mucosal bleeding is particularly common. Childhood epistaxis, a life-long history of easy bruising, bleeding with dental extraction, heavy menstrual bleeding or anemia attributed to excessive menstrual blood losses, or postpartum hemorrhage are all included in the spectrum of vWD symptomatology. Systemic bleeding disorders are often not considered in women with menorrhagia,
165 but clinical studies reveal that the prevalence if vWD is significant in individuals who present with this complaint.
166 Angiodysplasia of various vascular beds, particularly those in the gut, has been demonstrated in some patients with vWD and may be an important contributory factor in chronic gastrointestinal bleeding.
167 In type 1 vWD, symptoms usually become milder during pregnancy or with estrogen therapy, when the vWF and factor VIIIc levels rise significantly.
40 The disorder may also decrease in severity with advancing age. Bleeding manifestations tend to be more prominent in patients with more severe quantitative deficiency and in patients with qualitative (type 2) defects.
While eliciting the history of bleeding, one should bear in mind that women are often inaccurate assessors of whether their menstrual flow is normal or excessive. Supplemental questioning regarding the frequency of changes of menstrual protection, and questions regarding a history of iron deficiency anemia and transfusion, may be informative. Retrospective studies of women with vWD show that abnormal bleeding can often be traced to menarche.
168 Family history also may provide important clues to a diagnosis. The physician should specifically seek a history of the response to hemostatic challenge, such as dental extraction, tonsillectomy, surgical procedures, menstruation, peripartum hemorrhage, and transfusion. A model questionnaire with a scoring system has been published.
169 Coexistent conditions or drug effects can strongly influence the severity of bleeding symptoms. Risk of hemorrhage may increase in patients with concomitant liver disease, uremia, gastrointestinal ulcer, or angiodysplasia. Excessive bleeding in response to challenge with aspirin may also point toward a bleeding disorder, but this history is not specific for vWD. Valproic acid use can result in lower vWF activity levels, as may hypothyroidism and valvular heart disease. Conversely, oral contraception frequently ameliorates menorrhagia.
127
Type 1 von Willebrand Disease and “Low von Willebrand Factor”
Patients with bleeding attributed to decreased production of qualitatively normal vWF are classified as having type 1 vWD. Most patients with vWD (70% to 80%) fall into this category.
134 The majority of patients have a mildly symptomatic disorder, but bleeding may increase with physical trauma, surgery, or during menstruation. vWF levels are low, with concordant reduction of vWF:Ag and vWF:RCo (and vWF:CB, if that is measured). Factor VIII levels are generally equal to or higher than vWF levels, and all vWF multimers should be present if that analysis is performed as part of the patient evaluation. The diagnosis of type 1 vWD may be simpler in an individual with mucosal bleeding symptoms, a strong family history of similar symptoms, and quantitative vWF level (vWF:Ag and/or vWF:RCo) <30 IU/dl. Although not required for a diagnosis of type 1 vWD, the probability of finding linkage of the phenotype to the vWF gene in such an individual would be high, such that both therapeutic choices and genetic counseling would be straightforward.
Unfortunately, many factors make the diagnosis of type 1 vWD difficult.
144 Mild bleeding symptoms are common in hemostatically normal people (see
Chapter 45). In addition, the relative risk of bleeding for individuals with only modest reductions in vWF level appears to be minimal. Finally, because of the strong influence of blood group on vWF levels, there is a high prevalence of blood group O among individuals with mildly depressed vWF levels. These observations have led to the recent proposal of the concept of “low vWF”.
137,
144 The term “low vWF” could be applied to patients with vWF levels below the normal reference range but above some lower limit. A recently convened “expert group” brought together by the National Heart, Lung and Blood Institute is considering this proposal.
129 Studies of families of patients with a diagnosis of type 1 vWD suggest that linkage to the vWF gene is less often seen when the vWF level rises above 30 IU/dl, suggesting that this might be a candidate for the lower limit of the “low vWF” category.
145,
198 Empiric therapy to raise vWF levels in patients with “low vWF” at times of hemostatic challenge would be reasonable for patients with a history of bleeding, but the genetic counseling issues would clearly be different.
144On the other hand, two population-based studies have shown that “low vWF” causes increased bleeding.
199,
200 These latter studies would suggest that abnormally low ristocetin cofactor activity, and not the presence of a mutation, should primarily be used to identify bleeders.
Type 3 von Willebrand Disease
Type 3 vWD is the most severe form of vWD, because it results from complete failure of vWF synthesis. This bleeding disorder is generally diagnosed during infancy. Hematoma formation is common, epistaxis may be life-threatening, and hemarthrosis may occur as a result of the low factor VIII levels that are seen in this condition. Plasma from patients with type 3 vWD contains virtually no detectable vWF, and vWF is not present in either platelets or endothelial cells. Factor VIII level is generally in the range of 1 to 10 IU/dl, similar to that seen in moderate to mild hemophilia A. Multimeric analysis of the little vWF present yields variable results, in some cases revealing only small multimers.
193 Type 3 vWD is rare, with an estimated frequency of 1 in a million people.
201 Genetic analysis reveals either homozygous or compound (double) heterozygous defects of the vWF gene, with gene deletions, frame-shift mutations, missense mutations, or nonsense mutations. Careful laboratory evaluation of the parents of patients with type 3 disease may reveal mild quantitative deficiency of vWF. In a review of 117 obligate heterozygotes, the mean plasma vWF level was 45 IU/dl, with a range from 5 to 130 IU/dl.
144 Parents are frequently asymptomatic, consistent with the impression that type 3 disease is inherited as a recessive trait. Consanguinity is common in kindreds with this variant. In some patients with type 3 vWD, large gene deletions have been identified on one or both vWF alleles.
128,
202 Such patients are at increased risk of developing inhibitory alloantibodies to vWF after transfusion therapy.
202Because these patients lack vWF in their endothelial cells and platelets, there is no rise in vWF level in response to DDAVP. For replacement therapy, these patients require vWF-containing concentrates. This is usually provided in the form of specific intermediate-purity factor VIII concentrates that have been documented to contain intact vWF.
155 Because these patients possess a normal
factor VIII gene but are missing the vWF chaperone, posttransfusion factor VIII recovery and survival may be longer in a patient with type 3 vWD than in a patient with hemophilia A, owing to endogenous factor VIII production. Although life-threatening bleeds require immediate replacement of both vWF and factor VIII, replacement with vWF alone may be sufficient if therapy is begun 12 to 24 hours before elective surgery.
203
Qualitative Defects of von Willebrand Factor
Several subsets of patients with vWD differ substantially from those with the common quantitative deficiency states (
Table 53.6).
128 Classified as type 2 variants, these qualitative defects of vWF are less common, but they may represent up to 20% to 25% of cases of vWD. Type 2 forms of vWD are suspected when the severity of the patient’s symptoms seems in excess of the observed vWF and factor VIII levels, when there is discordant reduction between vWF antigen and vWF functional activity or factor VIII assay, or when there is concomitant vWF deficiency and thrombocytopenia. Discrimination between qualitatively normal and abnormal vWF can be difficult; however, when the ratio of vWF:RCo to vWF:Ag or vWF:CB to vWF:Ag is found to be <0.6, dysfunctional vWF should be considered. The sensitivity and specificity of such ratio screening depends to a large extent on the expertise and precision of the laboratory performing the assays, but the predictive value of ratios decreases when the level of vWF antigen is <20 IU/dl. Examining the structure of patient vWF may provide further definition of the nature of the qualitative defect. This can be demonstrated by vWF multimer analysis. If required, further information is provided by supplemental studies of vWF function, or via genetic analysis of the vWF gene (
Fig. 53.5).
126,
128,
193 Type 2 defects were initially classified using a Roman numeral system, but further understanding of the genetics of vWD led to the consolidation and reclassification into types 2A, 2B, 2M, and 2N,
128 based on the nature of the vWF functional defect. In addition to the well-defined variants described in this chapter, other forms of vWD continue to be described.
Type 2A von Willebrand Disease
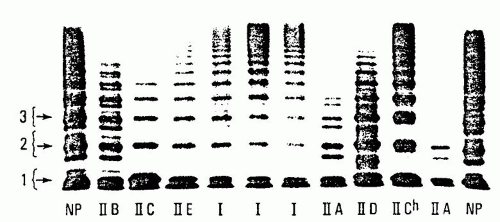
vWD with defective interaction of vWF with platelets due to deficiency of intermediate- and high-molecular-weight forms of vWF is classified as type 2A vWD.
128 This form of the disorder is generally inherited as an autosomal dominant trait,
204 and it accounts for the majority of patients with type 2 defects. Levels of factor VIIIc and vWF:Ag in the plasma may be normal or reduced. The vWF activity as assayed by either vWF:RCo or vWF:CB is significantly lower than vWF:Ag because of the absence of the larger multimers (which are more potent in their ability to interact with platelet GPIb and collagen). Analysis of vWF multimers reveals a relative reduction in intermediate- and high-molecularweight species.
127 Protein studies followed by genetic analysis of the basis of type 2A vWD revealed multiple mechanisms for the generation of this disorder.
205,
206,
207 In some patients, there is failure to synthesize full-length multimers (type 2A, subgroup 1). Mutations associated with impaired vWF multimerization have been localized to the vWFpp (formerly called type IIC), the region of the mature vWF protein associated with the amino terminus area of multimer formation (formerly called type IIE), and the carboxy terminus region that is involved in initial dimer formation (formerly called type IID). Subtle abnormalities uncovered by high-resolution multimer analysis or genetic evaluation can be used to distinguish these subtypes of type 2A vWD. The spectrum of multimers released into plasma after DDAVP administration would not be expected to improve significantly in these patients whose defects prevent normal multimer assembly.
A second group of type 2A patients has been described in whom there is excessive catabolism of fully multimerized vWF after vWF is released into the plasma. These patients demonstrate mutations in exon 28, encoding the A1 and A2 regions of vWF. These mutations allow increased proteolysis of multimerized vWF by ADAMTS13. Again, high-resolution multimer gel studies may be informative by demonstrating increased “satellite” band intensity, which accumulates as a result of excessive ADAMTS13 activity. Because intracellular vWF is protected from cleavage by ADAMTS13, these patients may demonstrate very transient
improvement in both vWF:RCo and multimer spectrum after DDAVP administration.
Inheritance of type 2A vWD is generally autosomal dominant, with the majority of cases caused by mutations clustered in the region of exon 28, which encodes the vWF A2 homologous repeat (
Fig. 53.5).
128 Rare recessive forms of type 2A vWD have been reported with some multimerization defects (formerly called types IIC and IID).
Because of the underlying structural abnormality of the vWF produced in these patients, neither stress nor pregnancy significantly increases the functional amount of vWF protein in the plasma. As noted above, the administration of DDAVP does not result in a significant rise in vWF:RCo in patients with type 2A vWD, in which the underlying mechanism is defective multimerization or transport, but a very short-lived correction may be observed in patients with mutations associated with increased vWF cleavage by ADAMTS13.
Type 2B von Willebrand Disease
Type 2B vWD is a paradoxical bleeding disorder, characterized by increased interaction of patient vWF with platelets which is demonstrated in vitro in the presence of low doses of ristocetin. Increased in vivo interaction of the larger multimers of type 2B vWF with platelets is thought to result from mutations that either allow increased access of platelets to the A1 loop of vWF or that stabilize that interaction.
127 This may in turn allow increased generation of vWF-platelet complexes, which are subsequently cleared from circulation, resulting in thrombocytopenia.
This bleeding disorder is characterized by deficient vWF function attributable to mild reduction of vWF:Ag, a somewhat more marked deficiency of vWF:RCo activity (and vWF:CB, if measured). Multimeric analysis reveals a deficiency of the highestmolecular-weight vWF multimers. Measurements of factor VIIIc are variable. Many patients with the type 2B variant have mild persistent thrombocytopenia.
208 The platelet count may fall further during physiologic stresses, pregnancy, in association with surgical procedures, or after the administration of DDAVP.
209Additional laboratory evaluation is required to support a diagnosis of type 2B vWD. In most cases, multimeric analysis reveals absence of higher-molecular-weight forms of plasma vWF, but distinction of type 2B from type 2A vWD is not possible by multimeric analysis alone. To show the distinct “gain in function” in type 2B disease, RIPA studies are done to reveal enhanced interaction of patient vWF and platelets with low doses of the drug. Finally, to differentiate the more common type 2B vWD from the rare platelet-type (pseudo) vWD, one should prove that the defect resides in vWF. This is done either through performing mixing studies of patient plasma with donor platelets
187 or by analysis of the patient’s vWF gene.
Type 2B vWD is inherited as an autosomal dominant trait. A small cluster of mutations within the portion of vWF exon 28 that encodes the vWF A1 domain that interacts with platelet GPIb accounts for all the cases of type 2B vWD that have been reported (
Fig. 53.5).
128Recent studies evaluating a large number of patients with type 2B vWD mutations revealed diversity in laboratory findings.
210 Not all patients had reduced vWF:RCo activity or abnormal multimeric analysis, and not all patients had thrombocytopenia.
210 Type 2B patients with normal multimers did not develop thrombocytopenia.
Type 2M von Willebrand Disease
The type 2M vWD variant is defined by diminished platelet-dependent vWF function that is not attributed to deficiency of vWF multimers.
128 Thus, these patients have an initial laboratory profile that in many ways is similar to that of patients with type 2A vWD, revealing variable deficiency of vWF:Ag but disproportionately decreased interaction of vWF with platelets in the presence of ristocetin as measured by vWF:RCo assay. Factor VIII level is proportionate to the vWF:Ag, and the platelet counts are normal. What differentiates type 2M vWD from the type 2A patients is that the vWF multimeric analysis is normal (and, if measured, the vWF:CB is usually similar to the vWF:Ag). Type 2M vWD is inherited as an autosomal dominant trait, and, when investigated, mutations have been found in the region of exon 28 that encodes the A1 domain of vWF. Unlike the type 2B mutations, 2M mutations cause impairment of the binding of vWF to the GP1b receptor and have been shown to be localized to an alternative area in the A1 domain (
Fig. 53.5).
196One case classified as a type 2M defect was described in which the vWF interaction with collagen was defective. In that case, a mutation in the A3 domain that contributes to the interaction of vWF with collagen was found.
189
Type 2N von Willebrand Disease
Mutations affecting the association of vWF with factor VIII can result in “autosomal hemophilia” in which factor VIII levels are significantly reduced relative to vWF.
128 Affected patients have factor VIII levels in the range of 5 to 30 IU/dl, attributed to mutations at the factor VIII-binding site near the amino terminus of the vWF subunit.
211 Indeed, genetic studies indicate that the majority of mutations underlying type 2N vWD involve vWF exons 18, 19, and 20, which encode the bulk of the factor VIII-binding domain of vWF. The other functions of vWF in patients with type 2N vWD are qualitatively normal, and thus vWF laboratory parameters (vWF:Ag, ristocetin cofactor activity, etc.) are usually normal (unless there is coinheritance of type 1 vWD). This variant was originally named vWD Normandy but has been renamed type 2N vWD in the revised classification.
128 The factor VIII-binding defect in these patients is inherited in an autosomal recessive manner, and thus affected patients must inherit 2N alleles from each of their two parents, or inherit type 1 vWD from one parent while inheriting a 2N allele from the other. Patients with factor VIII deficiency and a bleeding disorder that is not clearly transmitted as an X-linked disorder or who demonstrate an unexpectedly short in vivo survival of infused factor VIII should be evaluated for type 2N vWD.
212 Diagnosis requires performance of an assay that assesses the interaction of factor VIII with vWF,
191,
192 or genetic studies of the vWF gene. A survey of almost 400 unrelated patients with either hemophilia A or type 1 vWD indicated a prevalence of type 2N vWD in these patient populations of 3% and 1.5%, respectively.
213 Consideration of this diagnosis is important because both therapy and genetic counseling are distinct from that of mild hemophilia A.
Platelet-type (Pseudo) von Willebrand Disease
Platelet-type (pseudo) vWD resembles type 2B vWD in most respects, except that the basis for platelet-type vWD is a structural defect in platelet GP1b
214 rather than a defect of vWF (thus platelet-type [pseudo] vWD is a form of platelet dysfunction). However, as with type 2B vWD, a genetic defect results in increased interaction between GP1b and vWF. Phenotypically, platelet-type (pseudo) vWD is manifested by mild thrombocytopenia, a prolonged bleeding time, and variable deficiency of plasma vWF and factor VIIIc. The reductions in plasma vWF and factor VIIIc may be a result of the attachment of these proteins to platelets that are subsequently removed from the circulation. This “consumption” of vWF and platelets results in preferential loss of high-molecular-weight multimers from patient plasma.
214 In vitro platelet aggregation studies reveal platelet agglutination at unusually low concentrations of ristocetin. Spontaneous intravascular platelet clumping may also occur. Platelet-type (pseudo) vWF is inherited as an autosomal dominant trait in most families.
214,
215Distinction of platelet-type vWD from type 2B vWD is based on showing that the enhanced interaction of platelets with vWF is either platelet-based or resides in the plasma phase (type 2B vWD).
216 This can sometimes be demonstrated through low-dose ristocetin-based agglutination assays using mixtures of normal donor and patient samples, or genetic analysis can be used. To date, all cases of platelet-type vWD have been traced to genetic variations of platelet GPIb
α.217 These include missense mutations that alter the amino acid sequence from Gly233 to Met 239. This region of the protein adopts a
β-sheet conformation after interaction with vWF, and these mutations may function to stabilize this conformation.
218 In addition, a 27-bp gene deletion that encodes for deletion of proline 421 through serine 429 has been shown to underlie platelet-type vWD in one kindred. Thus, genetic analysis of the GPIb
α gene could confirm a diagnosis of platelet-type vWD in cases in which phenotypic studies were unsuccessful.