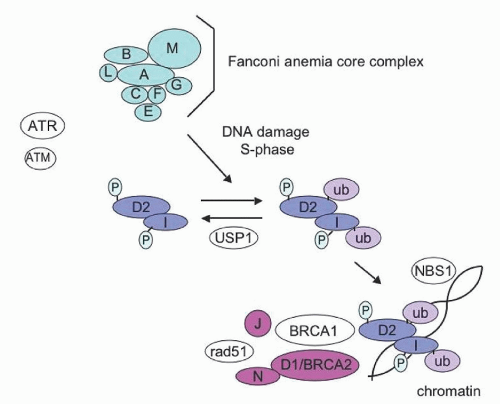
FA is an inherited chromosomal instability syndrome with a variable clinical presentation that includes congenital anomalies, progressive pancytopenia, and cancer susceptibility. Guido Fanconi first reported a familial syndrome of pancytopenia and congenital physical abnormalities in 1927.
3,
4 The diagnostic hallmark of FA is increased chromosomal breakage in response to DNA damaging agents such as mitomycin C (MMC) or diepoxybutane (DEB). Remarkable advances in the last few years have elucidated the molecular pathways disrupted in FA cells. Potential clinical implications of recent molecular studies will be discussed.
Clinical Features
FA should be considered in patients with congenital anomalies, aplastic anemia, or a family history of bone marrow failure or cancer susceptibility. The International Fanconi Anemia Registry (IFAR) analysis of 370 patients found that nearly 40% of patients had no reported physical findings.
5 Aplastic anemia or malignancy may be the presenting sign of the underlying diagnosis of FA in the absence of physical anomalies or prior family history. The manifestations of FA can vary between affected members of the same family, suggesting that additional genetic, epigenetic, or environmental factors likely influence the disease course.
A wide range of congenital anomalies has been reported in FA patients (reviewed in
Refs. 1,
6) (
Table 37.1). Skin pigmentary changes, short stature, and radial bone and thumb abnormalities are the most common manifestations associated with FA, although these may be seen in some of the other inherited bone marrow failure syndromes as well. Abnormalities of the skeletal, ocular, renal, genital, aural, gastrointestinal (GI), cardiac, and central nervous systems have also been reported with high frequency.
The hematologic complications of FA typically present within the first decade of life. Early manifestations include moderate single or bilineage cytopenia with red cell macrocytosis. Elevated levels of fetal hemoglobin (Hb F) help to distinguish inherited from acquired aplastic anemia. Marrow failure may range in severity from mild asymptomatic cytopenias to severe aplastic anemia. This is in contrast to other chromosomal instability syndromes, such as ataxia telangiectasia, or Bloom syndrome, which are not typically associated with bone marrow failure.
Patients with FA are at increased risk of developing myelodysplasia (MDS) or acute myeloid leukemia (AML).
7,
8 In a cohort study of 145 patients with FA, the cumulative incidence of AML reached 10% by age 24 years and plateaued thereafter.
8 Patients with FA are also at increased risk of developing solid tumors, particularly squamous cell carcinomas of the head and neck, skin, GI tract, and genital tract.
9 The cumulative incidence of solid tumors was low in younger patients but progressively increased with age starting at around 20 years of age, and reached 29% by age 48 years.
8 FA patients, particularly those who have received
androgen treatment, are also at increased risk of liver tumors. Treatment of malignancies is limited by the low tolerance of FA patients to chemotherapy and radiation. Myeloid malignancies in FA patients have been successfully treated with hematopoietic stem cell transplantation (HSCT), though patient numbers are small and follow-up is limited. The role of pretransplant chemotherapy is unclear.
10 For solid tumors, surgical resection is the mainstay of therapy. Regular and frequent surveillance for cancers is particularly important in this population and is discussed further in the section “Supportive Care.” Patients with the
FANCD1/BRCA2 subtype manifest an especially high rate of early onset AML and specific solid tumors (brain and Wilms) compared with other FA subtypes.
11,
12,
13Growth hormone deficiency has been observed in rare FA patients,
14 and treatment with growth hormone improved growth in a subset of these patients
15; its use is controversial because of a hypothetical risk of cancer or leukemia.
16 Additional endocrine disorders associated with FA include hypothyroidism with or without thyroid hormone binding globulin deficiency, abnormal glucose tolerance, and diabetes mellitus.
14,
17 Although FA is associated with short stature, endocrine evaluation is important to rule out treatable endocrine causes that might further contribute to poor growth.
There are currently 15 known FA subtypes (A, B, C, D1 [
BRCA2] , D2, E, F, G, I, J [
BRIP1, BACH1], L, M, N [
PALB2], O [
RAD51C], and P [
SLX4]). With the exception of subtype B, which is X-linked recessive, all the other FA subtypes follow an autosomal recessive pattern of inheritance.
18,
19 FA is found with similar frequencies in both genders and has no known ethnic restriction. The heterozygote carrier frequency has been recently reestimated at 1 in 181 in the United States and is higher in regions with founder effects such as Israel.
20 The mean age at diagnosis is generally reported to be between 7 and 9 years of age, with 75% of cases diagnosed between the ages of 4 and 14; however, FA has been diagnosed in neonates as well as in adults in their fifties.
Laboratory
Cell morphology on peripheral blood smear is typically unremarkable except for red cell macrocytosis and mild anisocytosis, though these are variably present. Thrombocytopenia or leukopenia typically precede anemia. Pancytopenia may be progressive over time. Erythrocyte macrocytosis and increased Hb F levels may be present even in the absence of cytopenias.
Bone marrow biopsy findings vary from normal cellularity to frank aplasia. Morphologic examination of the bone marrow aspirate may show dysplastic features, with nuclear-cytoplasmic maturation disynchrony. Dyserythropoiesis with multinucleate forms or nuclear fragmentation may be seen. Bone marrow aspirates should be sent for cytogenetic analysis, as FA patients are at high risk for malignant transformation. The appearance of cytogenetic clones of unclear clinical significance in the absence of morphologic evidence of MDS must be considered carefully, since cases of persistent or transient clonal abnormalities without apparent progression to leukemia have been reported.
23 At a minimum, patients with persistent unexplained changes in blood counts or new cytogenetic marrow abnormalities should be monitored closely with serial blood counts and a follow-up bone marrow examination to assess for possible malignant evolution. Amplification or gains of chromosome 3q26q29 were shown to correlate with increased risk of MDS and AML in one study.
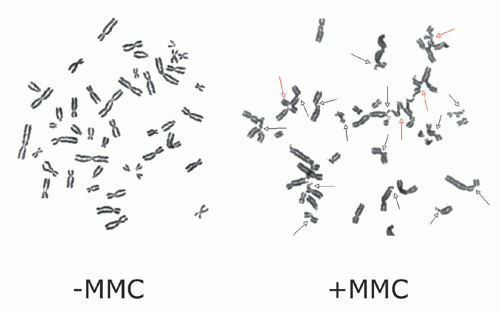
24The diagnosis of FA is based on the demonstration of increased chromosomal breakage in the presence of DNA cross-linking agents, such as MMC or DEB.
25,
26 (
Fig. 37.1) DEB is preferred in some centers since it is associated with less variability in chromosomal breakage among normal controls. The chromosomal breakage test is usually performed on metaphase spreads of peripheral blood lymphocytes cultured with MMC or DEB. A total of 50 metaphase cells are analyzed for chromosomal
breakage, including the formation of radial figures—a hallmark of this disease. Results are compared to a normal control and a positive control that have been run in parallel. Results are generally reported as aberrations per cell and the number of cells with breaks or radial forms. Increased spontaneous chromosomal breakage may be observed in some FA patients
12; nonetheless, the rate of breakage is markedly enhanced by exposure to MMC or DEB regardless of patient phenotype or severity of disease. Chromosomal breakage in response to MMC/DEB can also be assessed in fetal cells obtained for prenatal diagnosis by amniocentesis or chorionic villus sampling.
27,
28 If the disease-causing mutation is known for a given family, these assays can be used for prenatal diagnosis or preimplantation genetic diagnosis.
29 FA patient cells also exhibit cell cycle abnormalities with G2 phase prolongation and arrest by flow cytometry.
30,
31 Studies comparing results of DEB chromosomal breakage with cell cycle analyses of patients referred for FA testing showed close correlation between the results of these two tests.
32 Constitutive elevation of serum alpha-fetoprotein (AFP) has been reported in FA patients,
33 although variations in AFP levels determined by different methodologies and the lack of specificity has limited the diagnostic utility of this test so far.
34 FA heterozygous carriers cannot be reliably detected by testing for chromosomal breakage. Genetic mutation testing is available for the FA genes.
35Clonal somatic reversion to wild-type has been observed in a subset of cells from some FA patients. Increased chromosomal breakage may be limited to a subpopulation of lymphocytes in such patients. The reversion to normal cellular phenotype has been attributed to recombination or gene conversion events leading to selective growth advantage of the reverted cells
36,
37; somatic reversion has been reported in early hematopoietic lineages.
38 Patients with a high degree of wild-type mosaicism may be difficult to diagnose. In cases with a high degree of suspicion for FA and normal blood breakage analysis, the MMC/DEB test should be performed on skin fibroblasts, since somatic mosaicism has not been observed in fibroblasts.
Supportive Care
Cancer surveillance and education play an important role in the management of FA patients. Physicians should counsel patients regarding established behavioral and environmental risk factors associated with increased cancer risk. Since FA patients have a defect in DNA repair, imaging studies should minimize exposure to ionizing radiation. For example, when clinically feasible, it is reasonable to use MRI scans or ultrasound rather than CT scans for FA patients.
Because of the increased risk of MDS and leukemia in patients with bone marrow failure syndromes, frequent complete blood counts and annual bone marrow aspirates and biopsies with cytogenetic analysis are recommended, particularly since hematopoietic stem cell transplant outcomes are superior for patients transplanted prior to emergence of leukemia. FA patients are at risk for clonal cytogenetic abnormalities (reviewed in
Ref. 60). Assessment of the clinical significance of a cytogenetic clonal abnormality requires consideration of the specific clone, the number of chromosomal abnormalities, the presence of significant marrow dysplasia (some baseline mild dysplasia is common in FA), and whether there are concomitant progressive peripheral cytopenias.
23,
61 Chromosomal abnormalities common in AML, such as t(8:21), inv(16), or trisomy 8, have not been reported in FA.
23,
62 Amplifications of 3q are common in FA but rarely seen in AML. FA AML also had a higher frequency of 1q amplification, loss of part or all of chromosome 7, and gain of 13q and del 20q.
62,
63For patients on androgen therapy, regular physical examinations for liver size, liver ultrasound for masses or abnormalities every 6 to 12 months, and frequent liver enzyme tests are recommended. Annual examination by an otolaryngologist for leukoplakia or other signs of squamous cell carcinoma of the
oral cavity and oropharynx is important for patients with FA, DC, and those FA patients previously treated with bone marrow transplantation. Annual endoscopy can be considered in older FA and DC patients. Regular dental exams are important both for maintenance of oral hygiene and for detecting leukoplakia. Patients should be evaluated immediately for symptoms of pain in the mouth or throat, difficulty swallowing, changes in voice, anorexia, or weight loss. Suspicious lesions should be biopsied immediately, since early surgical excision is the mainstay of cancer therapy in FA patients. Annual gynecologic examinations, including Pap smears and HPV (human papilloma virus) exams, are recommended at puberty or after the age of 16. Counseling regarding sexual activity should be provided. Barrier methods of contraception may be particularly pertinent for the FA patients who are already at risk of cervical and vulvar malignancies. Regular breast exams are also recommended, although it is not clear what role mammography should play in cancer screening of these patients. HPV, which may be associated with an increased risk of squamous cell carcinoma of the head and neck, exhibits increased proliferation and epithelial cell expansion secondary to elevated levels of the viral E7 protein when the FA pathway has been disrupted.
64 Vaccination against HPV should be offered to all FA patients, males and females, in accordance with current guidelines.
Regular endocrinology evaluations are important, particularly in the pediatric population if the patient exhibits poor growth or delayed puberty. Hypothyroidism and diabetes are not infrequent in FA patients. A recent study reported one or more endocrine abnormalities in more than 70% of patients with FA.
17,
65 GI symptoms may present in FA patients, necessitating prompt referral to a gastroenterologist.