Andrew T. Kroger, Larry K. Pickering, Melinda Wharton, Alison Mawle, Alan R. Hinman, Walter A. Orenstein Keywords anthrax vaccine; antibody; assortant; BCG; Calmette-Guérin bacillus vaccine; cholera vaccine; coverage; diphtheria vaccine; effectiveness; efficacy; Haemophilus influenza vaccine; hepatitis A vaccine; hepatitis B vaccine; human papillomavirus vaccines; immunization; immunogenicity; influenza vaccine; Japanese encephalitis vaccine; measles vaccine; meningococcal vaccines; plague vaccine; pneumococcal vaccines; mumps vaccine; pertussis vaccine; polio vaccine; rabies vaccine; recombinant; rotavirus vaccines; rubella vaccine; smallpox vaccine; tetanus vaccine; toxoid; vaccine; varicella-zoster vaccine; yellow fever vaccine
Immunization
The two most effective means of preventing disease, disability, and death from infectious diseases have been sanitation and immunization. Both approaches antedated understanding of the germ theory of disease. Artificial induction of immunity began centuries ago with variolation, the practice of inoculating fluid from smallpox lesions into skin of susceptible persons. Although this technique usually produced mild illness without complications, spread of disease did occur, with occasional complications. In 1796, Jenner demonstrated that milkmaids who had contracted cowpox (vaccinia) were immune to smallpox. He inoculated the vesicular fluid from cowpox lesions into the skin of susceptible people and induced protection against smallpox, thus beginning the era of immunization.
Immunization, the act of artificially inducing immunity or providing protection from disease, can be active or passive. Active immunization consists of inducing the body to develop defenses against disease. This usually is accomplished by administration of vaccines or toxoids that stimulate the body’s immune system to produce antibodies or cell-mediated immunity, or both, which protects against the infectious agent.1 Passive immunization consists of providing temporary protection through administration of exogenously produced antibody. Two situations in which passive immunization commonly occurs are through transplacental transfer of antibodies to the fetus, which may provide protection against certain diseases for the first 3 to 6 months of life, and injection of immunoglobulins for specific preventive purposes. A more detailed description of the immune mechanisms involved follows.
Immunizing agents include vaccines, toxoids, and antibody-containing preparations from human or animal donors. Several important definitions are provided below.
The constituents of immunizing agents include the following:
2. Preservatives, stabilizers, antibiotics: these components of vaccines are used (1) to inhibit or prevent bacterial growth in viral culture or the final product (preservatives and antimicrobial agents) or (2) to stabilize (stabilizers) the antigen. They include materials such as mercurials (thimerosal), gelatin, and specific antimicrobial agents. Allergic reactions may occur if the recipient is sensitive to any of these additives. Preservatives are required for multidose vaccine formulations or vials to prevent bacterial or fungal growth, should they be introduced on repeated entry into the vial. Thimerosal, an ethyl mercury containing preservative, has been the major preservative used in vaccines around the world. A review of the mercury content of vaccines in the United States in 1999 indicated some children had received quantities of ethyl mercury from thimerosal in excess of some federal guidelines for methyl mercury. As a precautionary measure, thimerosal as a preservative was removed from most vaccines in the immunization schedule to the extent feasible.2 However, some of these vaccines may contain trace amounts (www.fda.gov/cber/vaccine/thimerosal.htm). Subsequent studies of potential adverse consequences of thimerosal have not demonstrated significant harm from its use in vaccines. It is likely had these data been available in 1999, the United States would not have made the decision to remove thimerosal from vaccines for children.3,4 Some vaccines for children contain other preservatives (e.g., 2-phenoxyethanol) or do not need a preservative because they are packaged in single-dose vials. Influenza vaccines in multidose vials used in adults and combined adult-type tetanus and diphtheria toxoids (Td) contain thimerosal as a preservative.5
3. Adjuvants: an aluminum salt is used in some vaccines to enhance the immune response to vaccines containing inactivated microorganisms or their products (e.g., toxoids and hepatitis B vaccine). One brand of human papillomavirus (HPV) vaccine contains an aluminum salt combined with monophosphoryl lipid A. Vaccines with such adjuvants should be injected deeply into muscle masses because SC or intracutaneous administration can cause local irritation, inflammation, granuloma formation, or necrosis.6 Oil-in-water adjuvants are used in some vaccines licensed outside the United States and have been documented to induce markedly enhanced immunogenicity for some vaccines, such as inactivated influenza H5N1 vaccines.7
Immunologic Basis of Vaccination
Two major approaches to active immunization have been used: use of live (attenuated) infectious agents or use of inactivated, or detoxified, agents or their extracts. For many diseases (including influenza, poliomyelitis, typhoid, and measles), both approaches have been used. Live-attenuated vaccines are believed to induce an immunologic response more similar to that resulting from natural infection than are killed vaccines. Inactivated or killed vaccines can consist of inactivated whole organisms (e.g., hepatitis A vaccine), detoxified exotoxin (e.g., diphtheria and tetanus toxoids), soluble capsular material either alone (e.g., pneumococcal polysaccharide), or covalently linked to carrier proteins (e.g., Haemophilus influenzae type b [Hib] conjugate vaccines), chemically purified components of the organism (e.g., acellular pertussis, inactivated influenza vaccines), or recombinant proteins (e.g., HPV, hepatitis B virus [HBV]).
Determinants of Immunogenicity
The immune system is complex, and antigen composition and presentation are critical for stimulation of the desired immune response. Immunogenicity is determined not only by the chemical and physical states of the antigen but also by the genetic characteristics of the responding individual, the physiologic condition of the individual (e.g., age, nutrition, gender, pregnancy status, stress, infections, immune status), and the manner in which the antigen is presented (route of administration, dose or doses and timing of doses, and presence of adjuvants).8,9
Live Versus Killed or Subunit Vaccines
Because the organisms in live vaccines multiply in the recipient, antigen production increases logarithmically until controlled by the immune response induced by the antigen. The live-attenuated viruses (e.g., measles, mumps, and rubella) generally are believed to confer lifelong protection in those who respond. By contrast, killed vaccines generally do not induce permanent immunity with one dose, requiring repeated vaccination and subsequent boosters to develop and maintain high levels of antibody (e.g., diphtheria, tetanus, rabies, typhoid). Exceptions to this general rule may include hepatitis B vaccine, for which long-term immunologic memory has been demonstrated for approximately 20 years after vaccination, and inactivated polio vaccine (IPV), for which the duration of immunity is unknown. Although the amount of antigen initially introduced is greater with inactivated vaccines, multiplication of organisms in the host results in a cumulatively greater antigenic input with live vaccines.
Most vaccines include protein antigens, which generate a T-lymphocyte–dependent immune response. This response induces immunologic memory, booster effects with repeat administration, and good immunogenicity in all age groups. However, purified bacterial capsular polysaccharide vaccines induce a T-lymphocyte–independent immune response, which does not lead to immune memory and cannot be boosted with repeated injections.10 Polysaccharide vaccines have poor immunogenicity in infants and young children. Covalent linkage of the polysaccharide to a carrier protein converts it from a T-lymphocyte–independent to a T-lymphocyte–dependent antigen (e.g., conjugated Hib, pneumococcal, and meningococcal vaccines), which produces a good immune response in infants and children.
Dose
The amount of antigen determines the immune response. Presentation of an insufficient amount of antigen may result in an absence of immune responsiveness. There is usually a dose-response curve relationship between antigen dose and peak response obtained beyond a threshold; however, responsiveness may reach a plateau, failing to increase beyond a certain level despite increasing doses of vaccine.
Adjuvants
The immune response to some inactivated vaccines or toxoids can be enhanced by addition of adjuvants, such as aluminum salts (either alone or in combination with monophosphoryl lipid A). Adjuvants are particularly useful with inactivated products, such as diphtheria and tetanus toxoids, acellular pertussis vaccines (DTaP), and hepatitis B vaccine. The mechanism of enhancement of antigenicity by adjuvants is not well defined; however, it is increasingly clear that adjuvants activate the innate immune system through pathogen-associated molecular patterns (PAMPs). Although aluminum salts alone or with monophosphoryl lipid A are currently the only licensed adjuvants for use in humans in the United States, other adjuvants based on oil-in-water emulsion of squalene have shown promising results.11
Route of Administration
The route of administration can determine the nature of the immune response to a vaccine or toxoid. IM and SC delivery results in a predominantly IgG response. Oral (e.g., rotavirus vaccine and typhoid vaccine Ty21a) or nasal (e.g., live-attenuated influenza vaccine) vaccination is more likely to result in production of local IgA compared with IM injection, although systemic IgG also is induced. The immunogenicity of some vaccines is reduced when not given by the recommended route. For example, administration of hepatitis B vaccine subcutaneously into the fatty tissue of the buttock was associated with substantially lower seroconversion rates than injection intramuscularly into the deltoid muscle.12
Most vaccines are administered either intramuscularly or subcutaneously. One formulation of influenza vaccine is administered intradermally. Administration by this route reduces the amount of antigen per dose, thereby decreasing pressure on production capacity and, potentially, vaccine cost.13
Age
The immune response to a vaccine varies with age. Although children and young adults usually respond well to all vaccines, differences in response capability exist during early infancy and older age. The presence of high levels of passively acquired maternal antibody in the first few months of life impairs the initial immune response to some killed vaccines (e.g., hepatitis A vaccine,14 diphtheria toxoid) and many live vaccines (e.g., measles). Prematurely born infants of low birth weight should be immunized at the usual chronologic age in most cases. Infants with birth weights less than 2000 g may require modification of the timing of hepatitis B immunoprophylaxis, depending on maternal hepatitis B surface antigen (HBsAg) status.15 Some studies suggest a reduced immune response in very-low-birth-weight infants (<1500 g) immunized by the usual schedule; however, antibody concentrations achieved usually are protective. In adults 40 years of age and older, the response to antigenic stimulation may be diminished (e.g., influenza, hepatitis B vaccines).
Components of the Immune Response
The immune response traditionally is divided into two components: the innate immune response, which is rapid, nonspecific, and serves as an immediate first line of defense against an infection, and the adaptive immune response, which develops over a matter of days, is specific for the foreign antigen, and results in long-term immune memory. The latter protects the host against subsequent challenge with the same or immunologically similar pathogens and is the underlying principle of vaccination. The innate immune response is mediated by natural killer (NK) cells, which recognize and kill virally infected cells; by complement, which is activated by components of bacterial cell walls; and by phagocytes, including macrophages and dendritic cells (DCs), which ingest microorganisms and foreign particulates.16 The adaptive immune response relies on antigen-presenting cells (APCs), such as DCs, for activation and is mediated by T and B lymphocytes. T lymphocytes can be divided into CD4 (helper) and CD8 (cytotoxic) lymphocytes and are responsible for cell-mediated immune responses. CD4 helper T lymphocytes can be further subdivided into Th1 lymphocytes, which predominantly lead to cell-mediated responses, and Th2 lymphocytes, which predominantly lead to humoral responses. B lymphocytes produce antibody specific for the immunizing agent and require CD4–T-lymphocyte help. Interactions between APCs, helper T lymphocytes, and B lymphocytes involve class II major histocompatibility (MHC) antigens, whereas interactions between cytotoxic T lymphocytes and their target involve MHC class I antigens.17 Soluble mediators or cytokines are secreted by all cell types and serve as activation and differentiation factors for different cell lineages. These include interleukins, interferons, and others.18 A further class of CD4 T-lymphocytes (Treg) plays an essential role in the regulation of the adaptive immune response.19
The innate immune response is able to respond differently to different types of pathogens, and these differential responses help determine the nature of the subsequent adaptive response.20 Pattern recognition receptors (PRRs) encoded in the germline recognize PAMPs, such as Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and others, all of which contribute to immune activation by inducing proinflammatory cytokines, which in turn modulate the adaptive immune response. As alluded to earlier, this has significant implications for adjuvant development.21,22
Mobilization of the Adaptive Immune Response
Upon exposure to an infectious organism or a vaccine, the innate immune system is mobilized through APC recognition of PAMPs that are present either in the organism or in the adjuvant. Activated APCs (macrophages and DCs) secrete proinflammatory cytokines and chemokines, which recruit other leukocytes to the site of infection. When activated, DCs migrate to the draining lymph nodes, where they interact with T lymphocytes through the MHC-peptide complex.23 Once the organism or antigen is internalized, it is killed and broken down into peptides. These peptides are transported to the cell surface through membrane trafficking and bind to MHC class I or class II molecules. MHC class I molecules are able to bind peptides that are 8 to 10 amino acids in length, whereas MHC class II molecules are more permissive, binding peptides of 13 amino acids and greater.24
The first step in the induction of a T-lymphocyte–dependent antibody response is the activation of naïve CD4 helper T lymphocytes by presentation of an antigen by phagocytes or dendritic cells. The T-lymphocyte receptor recognizes the MHC-peptide complex, and this recognition triggers secretion of cytokines, which stimulate maturation of naïve helper T lymphocytes. In the presence of interleukin-12 (IL-12), Th1 lymphocytes will differentiate, and these in turn will secrete IL-2 and interferon-γ. In the presence of IL-4, Th2 lymphocytes will differentiate and secrete IL-4 and IL-5. These two cytokines are essential for the differentiation and maturation of B lymphocytes into antibody-secreting plasma cells.
Naïve B lymphocytes recognize a specific antigenic epitope on native antigen through the immunoglobulin receptor on their surface but are unable to differentiate into antibody-secreting lymphocytes without T-lymphocyte help. A given B lymphocyte can only be activated by a T lymphocyte responding to the same antigen, though not necessarily to the same epitope. A helper T lymphocyte will recognize the MHC class II complex on the surface of the B cell and deliver a signal for B-lymphocyte differentiation. This leads to B-lymphocyte proliferation and maturation in a clonal manner. Class switching (from IgM to IgG and IgA) and affinity maturation occur, and antigen-specific plasma cells develop. However, not all B lymphocytes become plasma cells. Some mature into memory B cells, which are long lived and form the basis of the rapid secondary response on the next encounter with the pathogen.25 Although the mechanism of maintenance of these cells is not clear, the ability to mount a strong secondary response after many years argues for a homeostatic mechanism that regulates these cells. The antibodies formed after vaccination express a variety of antigen-binding specificities (i.e., recognize different structures on a complex multideterminant antigen), reflecting the sum of the large number of individual clonal B-lymphocyte responses that make up an antibody response.
Antibodies mediate protection through a variety of mechanisms. They may inactivate soluble toxic protein products of bacteria (antitoxins), facilitate intracellular digestion of bacteria by phagocytes (opsonization), interact with components of serum complement to damage the bacterial membrane with resultant bacteriolysis (lysins), prevent infectious virus from infecting cells (neutralizing antibodies), or interact with components of the bacterial surface to prevent adhesion to mucosal surfaces (anti-adhesins). Antibodies cannot readily reach intracellular sites of infection, the sites of viral and some bacterial replication. However, antibodies are effective against many viral diseases by interacting with virus before initial intracellular penetration occurs and by preventing locally replicating virus from disseminating from the site of entry to an important target organ, as in the spread of poliovirus from the intestine to the central nervous system or rabies from a puncture wound to peripheral neural tissue.
Virally infected cells can be killed by cytotoxic CD8 T lymphocytes. As the virus replicates in a cell, viral proteins are processed and presented on the cell surface as an MHC class I–peptide complex, which is then recognized by cytotoxic T lymphocytes. Cells infected with intracellular bacteria, such as Mycobacterium leprae, are recognized and killed in the same way.
Unanticipated Responses
Independent of antibody production, stimulation of the immune system by vaccination may, on occasion, elicit a hypersensitivity response. Killed measles vaccine, used in the United States between 1963 and 1967, induced incomplete humoral immunity and cell-mediated hypersensitivity, resulting in development of a syndrome of atypical measles in some children on subsequent challenge.26 In addition, some antibodies produced may not be protective but block the reaction of protective antibodies with antigens, inhibiting the body’s defenses. Some vaccines may induce immunologic tolerance that results in blunting of the immune response on subsequent exposure to the antigen (e.g., meningococcal polysaccharide vaccine).27 Concerns have been raised that immunizations might induce chronic allergic, autoimmune disorders, or autism. However, careful reviews of both the possible biologic mechanisms and epidemiologic evidence generally have failed to confirm vaccines as causes of these disorders.28 Concerns also have been raised that the number of antigens in the current vaccine schedule might overwhelm an infant’s immune system, leading to chronic diseases and predisposing to other serious infections. As a result of removal of whole-cell pertussis vaccine and smallpox vaccine from the current immunization schedule, the number of immunogenic proteins and polysaccharides a child is exposed to today is actually smaller than in the past. Estimates suggest that an infant is capable of responding to 10,000 vaccines simultaneously.29 The Institute of Medicine (IOM) concluded that available evidence favored rejection of a causal relationship between vaccines and increased risk for infections and type 1 diabetes mellitus. The evidence was insufficient to accept or reject a causal relationship between vaccines and allergic disorders, particularly asthma.30 A subsequent epidemiologic study failed to show an association between vaccines and asthma.31
Temporal Course of the Immune Response
On first exposure to a vaccine, a primary response is induced, and a protective immune response will develop in about 2 weeks. Circulating antibodies do not usually appear for 7 to 10 days, and the immunoglobulin class of the response changes over this period of time. Early-appearing antibodies are usually of the IgM class and of low affinity; late-appearing antibodies are usually of the IgG class and display a high affinity. IgM antibodies may fix complement, making lysis and phagocytosis possible. As the titer of IgG rises during the second week (or later) after immunogenic stimulation, the IgM titer falls. IgG antibodies are produced in large amounts and function in the neutralization, precipitation, and fixation of complement. The antibody titer frequently reaches a peak in about 2 to 6 weeks and then falls gradually. The switch from IgM synthesis to predominantly IgG synthesis in B lymphocytes is mediated by T-lymphocyte help. Uncommonly, people may not respond to a vaccine, experiencing a primary vaccine failure. This may be due to a genetic inability to respond to vaccine, but other factors are involved. For example, almost all children who do not respond immunologically to the first dose of measles-mumps-rubella (MMR) vaccine will acquire measles immunity after a second dose.32
After a second exposure to the same antigen, a heightened humoral or cell-mediated response, an anamnestic response, is observed. These secondary responses occur sooner than the primary response, usually within 4 to 5 days, and depend on a marked proliferation of antibody-producing cells or effector T lymphocytes. The secondary response depends on immunologic memory after the first exposure mediated by both T and B lymphocytes. Infection with measles or varicella vaccine strains has been shown to evoke a cell-mediated as well as humoral response.
Many pathogens replicate at mucosal surfaces before host invasion and may induce secretory IgA along the respiratory and gastrointestinal mucous membranes and at other localized sites (e.g., polio, rubella, influenza, rotavirus). IgA antibodies are efficient at virus neutralization (e.g., polio), fix complement through the alternative pathway (e.g., cholera), prevent adsorption of organisms to the intestinal wall (e.g., Escherichia coli, cholera), and can lyse gram-negative bacteria (with the aid of both complement and lysozyme).33 Current parenteral, especially inactivated, vaccines rarely induce high levels of secretory IgA antibodies.
Measurement of the Immune Response
Response to vaccines is often gauged by measuring the appearance and concentration of specific serum antibodies.34 For some viral vaccines, such as those for measles and rubella, the presence of circulating antibodies correlates with clinical protection. Although this has served as a dependable indicator of immunity, seroconversion measures only the humoral parameter of the immune response. Secondary vaccine failure occurs when an individual who previously had developed an adequate immune response loses measurable antibodies over time. This waning immunity can be attributed to a loss of long-lived memory B or T cells in the absence of repeated exposure to the pathogen. Evaluating persistence of antibody has been used to determine the duration of vaccine-induced immunity. However, the absence of measurable antibody may not mean that the individual is unprotected. Although a fall in titer occurs, on revaccination or challenge, a rapid secondary response is observed in IgG antibodies, with little or no detectable IgM response, suggesting persistent protection. With some vaccines and toxoids, the mere presence of antibodies is not sufficient to ensure clinical protection, but rather, a minimal circulating level of antibody is required (e.g., 0.01 IU/mL of tetanus antitoxin). Functional antibody is important in assessing immunity to bacterial polysaccharide vaccines. Opsonophagocytic activity is considered the assay of choice for monitoring vaccine response35 because the vaccines also induce nonfunctional antibodies that are detected in standard enzyme immunosorbent assay (EIA), although the EIA can be used as a proxy. Some immune responses may not in themselves confer immunity but may be sufficiently associated with protection that they remain useful proxy measures of protective immunity (e.g., vibriocidal serum antibodies in cholera). The measurement of cell-mediated immunity, which would be helpful in assessing the degree of ongoing protection in many circumstances, usually is limited to research laboratories and to only a few vaccines.
Vaccine Development
Most vaccines in use today have been developed by conventional techniques.36 For live-attenuated viral vaccines, organisms are repeatedly passaged in various tissue culture cell lines to reduce virulent properties while maintaining immunogenicity. Inactivated vaccines usually have been developed by growing microorganisms, followed by concentration, purification, and inactivation, not necessarily in that order. Component vaccines usually are derived from chemical separation of the needed component from the parent organism.
Future vaccines are likely to be derived from new methods of biotechnology, especially recombinant techniques. Currently available hepatitis B vaccines were developed by cloning the HBsAg gene into yeast, leading to synthesis of HBsAg within the yeast cell. Other new approaches for producing vaccines include live vectors, in which one or more genes encoding critical determinants of immunity from pathogenic microorganisms are inserted into the genome of the vector, followed by the administration of the vector as a component of the vaccine. These vectors may include viruses, such as poxviruses (vaccinia or canarypox), or bacteria, such as Salmonella or Calmette-Guérin bacillus (BCG). Other newer techniques include microencapsulation of critical antigens in polymers, which can lead to sustained release or pulse release over prolonged periods, mimicking the effect of multiple injections of an antigen over a several-month interval. New technologies also include use of nucleic acids, which encode critical antigens. Injection of the DNA, combined with administration of a protein at a later point in time, leads to production of antigen without risk for producing whole infectious organisms. Live-attenuated influenza vaccine was developed using genetic reassortment of the genes encoding two of the surface glycoproteins from wild virus isolates with six other genes contributed from a cold-adapted, temperature-sensitive influenza strain. Similar techniques were used to develop bovine rotavirus vaccines.37 Last, newer technologies focus on the development of adjuvants to help stimulate the immune response.38
General Principles of Immunization
Introduction and widespread use of vaccines resulted in global eradication of smallpox, elimination of poliomyelitis caused by wild viruses in the United States and most of the countries of the world, and dramatic reductions in the incidence rates of other diseases (Tables 321-1 and 321-2). Measles and rubella are no longer considered endemic in the United States.39,40 Diphtheria and rubella have been reduced by greater than 90% in developed countries and, if global vaccination efforts can be sustained, may eventually be eliminated from many countries.41 The World Health Assembly had established a goal to eradicate polio from the world by the end of 2000.42 Although that goal was not achieved, by the end of 2012, only three countries in the world had never interrupted wild poliovirus transmission, although several others in Africa had been reinfected (www.polioeradication.org).43 The last case of polio caused by wild virus in the Western Hemisphere was in 1991; three of the six regions of the World Health Organization (WHO)—American, European, and Western Pacific—have been certified free of wild polio virus.44–46 Global use of hepatitis B vaccine in infants may have an impact comparable to that of other vaccines in childhood. Hib vaccines have only recently come into widespread use, but disease incidence has been reduced markedly in many developed countries.47–50
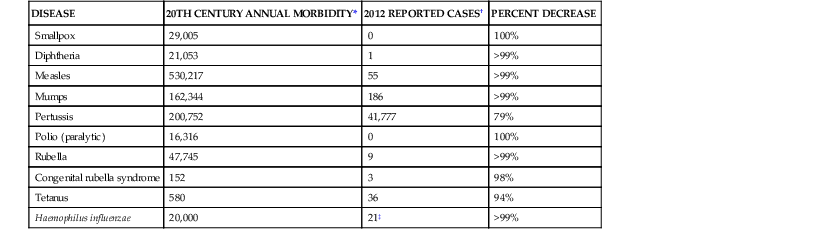
TABLE 321-1
Representative 20th-Century Morbidity Cases in 2012 and Change
| DISEASE | 20TH CENTURY ANNUAL MORBIDITY* | 2012 REPORTED CASES† | PERCENT DECREASE |
| Smallpox | 29,005 | 0 | 100% |
| Diphtheria | 21,053 | 1 | >99% |
| Measles | 530,217 | 55 | >99% |
| Mumps | 162,344 | 186 | >99% |
| Pertussis | 200,752 | 41,777 | 79% |
| Polio (paralytic) | 16,316 | 0 | 100% |
| Rubella | 47,745 | 9 | >99% |
| Congenital rubella syndrome | 152 | 3 | 98% |
| Tetanus | 580 | 36 | 94% |
| Haemophilus influenzae | 20,000 | 21‡ | >99% |
* From Roush SW, Murphy TV; Vaccine-Preventable Disease Table Working Group. Historical comparisons of morbidity and mortality for vaccine-preventable diseases in the United States. JAMA. 2007;298;2155-2163.
† From Centers for Disease Control and Prevention. Notifiable diseases and mortality tables. MMWR Morb Mortal Wkly Rep. 2013;61:ND-719-ND-731 [provisional week-52 data].
‡ Haemophilus influenza type b (Hib) at <5 years of age. An additional 14 cases of Hib are estimated to have occurred among the 227 reports of Hib (<5 years of age) with unknown serotype.
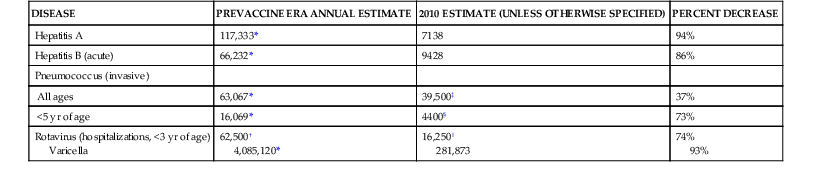
TABLE 321-2
Representative 20th-Century Morbidity Cases in 2012 and Change
| DISEASE | PREVACCINE ERA ANNUAL ESTIMATE | 2010 ESTIMATE (UNLESS OTHERWISE SPECIFIED) | PERCENT DECREASE |
| Hepatitis A | 117,333* | 7138 | 94% |
| Hepatitis B (acute) | 66,232* | 9428 | 86% |
| Pneumococcus (invasive) | |||
| All ages | 63,067* | 39,500‡ | 37% |
| <5 yr of age | 16,069* | 4400§ | 73% |
| Rotavirus (hospitalizations, <3 yr of age) Varicella | 62,500† 4,085,120* | 16,250‖ 281,873 | 74% 93% |
* From Roush SW, Murphy TV; Vaccine-Preventable Disease Table Working Group. Historical comparisons of morbidity and mortality for vaccine-preventable diseases in the United States. JAMA. 2007; 298;2155-2163.
† From Centers for Disease Control and Prevention. Prevention of rotavirus gastroenteritis among infants and children: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2009;58(RR-02):1-25.
‡ From Centers for Disease Control and Prevention. Active Bacterial Core Surveillance (ABCs) Report: Emerging Infectious Program Network: Streptococcus pneumoniae, 2009. Available at http://www.cdc.gov/abcs/reports-findings/survreports/spneu09.html.
§ From 2010 (provisional) Active Bacterial Core Surveillance.
‖ From New Vaccine Surveillance Network 2011 data (unpublished).
Pneumococcal conjugate vaccines have had a marked impact on invasive pneumococcal disease in countries where they have been used widely in children.51,52 Decreases in disease were observed not only in children but also in adults, who presumably are not being exposed to infectious children because the latter have had pneumococcal carriage eliminated by vaccination.
Modern vaccines are safe and generally effective. Each vaccine is associated with some adverse effects, which are usually mild, and only rarely life threatening. No vaccine is 100% effective. Consequently, some persons who have received a complete vaccine or toxoid series may acquire disease after exposure. The effectiveness of vaccines recommended for universal use in children is well defined, with most vaccines protecting more than 80% of recipients after a primary series.
For routinely recommended vaccines, there is no need to restart a series if the interval between doses is delayed. In most studies acellular pertussis vaccines range in efficacy from 71% to 89% during the first few years after vaccination.53,54,55 Varicella vaccine is 95% or more effective against severe varicella but is less effective against varicella of any severity.56,57 With some vaccines, antibody may wane, but immunologic memory is sufficient to prevent disease if exposed (e.g., hepatitis B).58 However, for some diseases with short incubation periods (e.g., meningococcal disease) waning antibody after vaccination is associated with waning protection. This waning has occurred with meningococcal conjugate vaccines, resulting in the need for modification of the originally recommended vaccine schedule with the addition of a second dose.59 Another example of loss of durability has occurred with the Tdap (tetanus, diphtheria, and acellular pertussis) and DTaP (diphtheria and tetanus toxoids and acellular pertussis) vaccine vaccines, in which protection begins to wane a few years after administration.60–62 In 2012 reductions of 95% or more from baseline 20th-century morbidity have been reported in the United States for smallpox, diphtheria, tetanus, polio, measles, mumps, and rubella. Similar reductions, based on historic estimates, have been achieved for congenital rubella syndrome and Hib invasive disease.63 Despite these successes, cases of measles and pertussis continue to occur in the United States (see Table 321-1).
Although high efficacy of each of these vaccines is apparent, there has been substantial controversy about reported adverse events temporally associated with vaccination. Because of these controversies, the IOM reviewed available information and between 1991 and 2013 published multiple reports.64–67 In the 1991 and 1994 studies, the IOM favored rejection of a causal relationship between combined diphtheria and tetanus toxoids (DT) and encephalopathy and between conjugate Hib vaccines and early-onset Hib disease. The IOM also concluded that the evidence favored a causal relationship between MMR and thrombocytopenia, between rubella vaccine and acute arthritis, between DT and brachial neuritis, and between a variety of vaccines and anaphylaxis. In 2004 the IOM reported the relationships between a variety of disorders and vaccines (www.iom.edu/Activities/PublicHealth/ImmunizationSafety.aspx).67,68 The IOM panel concluded that evidence did not support a relationship between MMR or thimerosal and autism, between multiple immunizations and heterologous infections, between multiple immunizations and type 1 diabetes, or between hepatitis B vaccine and incident or relapsed multiple sclerosis. In 2011 the IOM looked at the relationship of vaccines with many conditions that are reported after vaccination and, in most cases, found no evidence to support such associations. The IOM specifically found evidence to support rejection of an association between MMR vaccine and autism. Likewise, they found evidence to reject an association between inactivated influenza vaccine and asthma. In 2013 the IOM studied the impact of giving multiple vaccines to an individual in accordance with Advisory Committee on Immunization Practices (ACIP [of the Centers for Disease Control and Prevention {CDC}]), American Academy of Pediatrics (AAP), and American Academy of Family Physicians (AAFP) recommendations and found no evidence of a safety concern with adhering to the childhood schedule.69
Development of vaccines consists of four phases. Initial studies typically are conducted in animal models to demonstrate protection (or at least production of antibodies) and relative safety. Then limited numbers of doses are administered to humans to demonstrate antibody production and safety (phase I). After this phase, clinical trials in humans are conducted in a limited number of people to select optimal vaccine schedules and to demonstrate further safety (phase II). Larger trials are conducted to demonstrate efficacy (phase III). Because of their limited size, these field trials only can be expected to detect adverse events that occur relatively frequently (1 per 1000 doses or higher). After clinical trials, licensure may be sought. In the United States, vaccine production is regulated by the Center for Biologics Evaluation and Research (CBER) of the Food and Drug Administration (FDA). Only after a vaccine is found to be safe and effective is it licensed for use. Postmarketing surveillance (phase IV) is necessary to detect rare adverse events associated with vaccination and to monitor safety of vaccination practices, such as simultaneous immunization.
There is no direct evidence of risk to the fetus when pregnant women are given routinely recommended inactivated vaccines. The benefit of inactivated influenza vaccine to the pregnant mother and the fetus outweighs any risk of vaccination to the mother or the fetus. Some vaccines are recommended for pregnant women to provide passive immunity to their fetuses so that when the child is born, the child is protected before the time active immunity can be induced through direct vaccination of the infant. Thus, Tdap vaccine is recommended during each pregnancy.5,70 Most live-virus vaccines induce viremia, which at least theoretically could result in infection of the fetus, so live-virus vaccines are not administered to pregnant women except in unusual circumstances, when potential benefit clearly outweighs the risk.
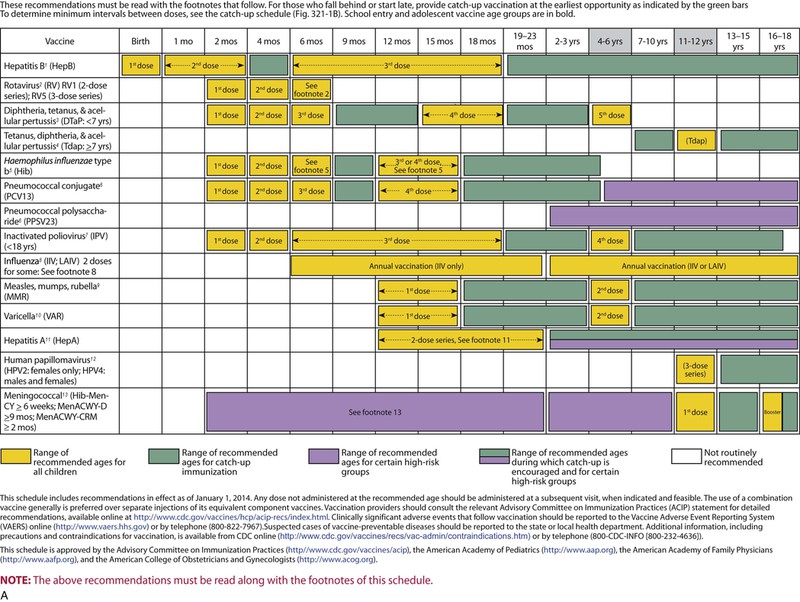
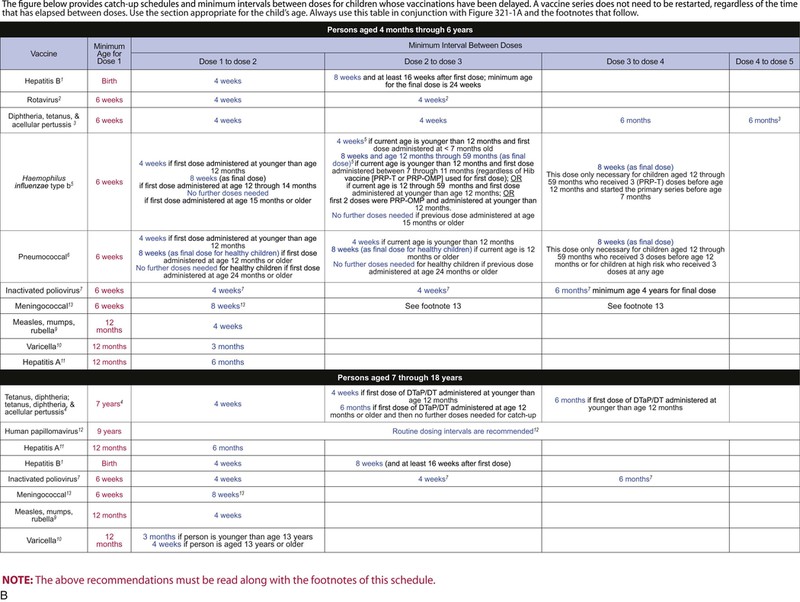
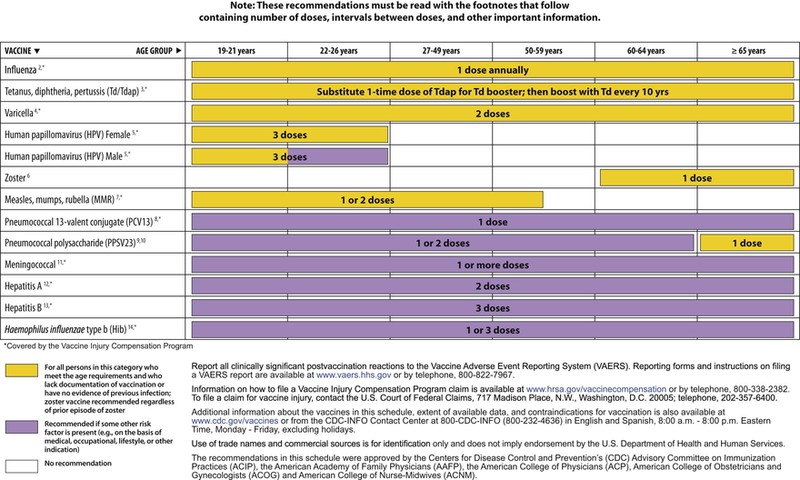
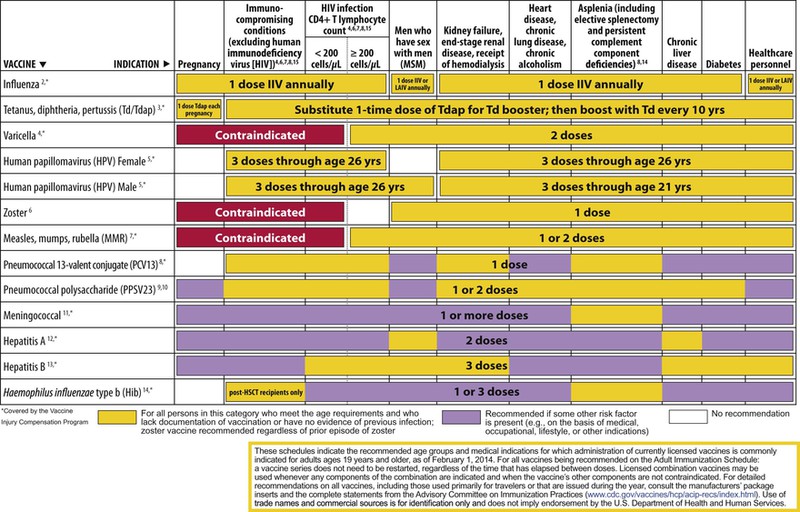
The decision to administer a vaccine involves assessment of risks of disease, benefits of vaccination, and risks associated with vaccination. The relative balance of risks and benefits may change over time; consequently, continuing assessment of vaccines is essential. Recommendations for vaccine use are developed by several different bodies: ACIP develops recommendations for vaccines for children, adolescents, and adults in the civilian population in conjunction with professional societies. These recommendations are updated annually and are available at www.cdc.gov/vaccines/schedules/hcp/index.html. Since 2011, the ACIP process for making vaccine recommendations has included a careful evaluation of the strength of the evidence supporting recommendations, which is known as GRADE (Grading of Recommendations, Assessment, Development, and Evaluation; www.cdc.gov/vaccines/acip/recs/GRADE/table-refs.html). The Committee on Infectious Diseases (COID) of the AAP (the “ Red Book “ committee) develops recommendations for vaccine use in infants, children, and adolescents.71 Since 1995, ACIP, the AAP, and the AAFP have collaborated to issue a harmonized childhood immunization schedule, which is updated annually (Fig. 321-1, A and B).72 ACIP also annually issues an adult immunization schedule in two parts: (1) recommendations based on age group and (2) recommendations based on underlying medical conditions (Figs. 321-2 to 321-4), which can be found at www.cdc.gov/vaccines/schedules/hcp/adult.html. The Adult Immunization Schedule for 2014 was harmonized with the AAFP, the American College of Obstetricians and Gynecologists, the American College of Physicians, and the American College of Nurse-Midwives.





Currently Available Immunizing Agents
Tables 321-3 and 321-4 list currently licensed immunizing agents and immunoglobulins. This section presents brief information about most immunizing agents, primary indications for use, relative efficacy, number and spacing of doses required, known adverse effects, and precautions and contraindications for use. Package inserts and specific references and recommendations should be consulted for more detailed information. In addition to these licensed products, several other vaccines are under development and may become available.
TABLE 321-3
Currently Available Vaccines and Toxoids and Year Licensed*
| PRODUCT | YEAR LICENSED |
| Anthrax vaccine adsorbed | 1972 |
| Calmette-Guérin bacillus vaccine; live, attenuated | 1950 |
| Diphtheria and tetanus toxoids and acellular pertussis vaccine | 1991 |
| Diphtheria and tetanus toxoids adsorbed (pediatric use, DT) | 1949 |
| Diphtheria and tetanus toxoids and acellular pertussis vaccine absorbed, Haemophilus B conjugate vaccine, and inactivated polio vaccine combined | 2008 |
| Diphtheria and tetanus toxoids and acellular pertussis vaccine absorbed and inactivated polio vaccine combined | 2008 |
| Diphtheria and tetanus toxoids and acellular pertussis adsorbed, hepatitis B (recombinant), and inactivated poliovirus vaccine combined | 2002 |
| Haemophilus influenzae type B conjugate vaccine | 1987 |
| Hepatitis A vaccine | 1995 |
| Hepatitis A inactivated and hepatitis B (recombinant) vaccine | 2001 |
| Hepatitis B recombinant vaccine | 1987 |
| Hepatitis B recombinant vaccine and Haemophilus influenzae type b conjugate vaccine | 1996 |
| Human papillomavirus vaccine | 2006 |
| Influenza virus vaccine (cell culture) | 2013 |
| Influenza virus vaccine (inactivated) | 1945 |
| Influenza virus vaccine; live, attenuated, intranasal | 2003 |
| Japanese encephalitis vaccine | 2009 |
| Measles virus vaccine; live, attenuated | 1963 |
| Measles, mumps, rubella, varicella; live, attenuated | 2005 |
| Measles, mumps, and rubella virus vaccine; live, attenuated | 1971 |
| Meningococcal groups C and Y and Haemophilus b tetanus toxoid conjugate vaccine | 2012 |
| Meningococcal polysaccharide vaccine, groups A, C, Y, W combined | 1981 |
| Meningococcal polysaccharide (serogroups A, C, Y, and W) conjugated to diphtheria toxoid | 2005 |
| Mumps virus vaccine, live, attenuated | 1967 |
| Pneumococcal conjugate vaccine (13-valent) | 2010 |
| Pneumococcal polysaccharide vaccine (23-valent) | 1983 |
| Poliomyelitis vaccine (inactivated, enhanced potency) | 1987 |
| Rabies vaccine (human diploid) | 1980 |
| Rotavirus vaccine, live, attenuated | 2006 |
| Rubella virus vaccine, live, attenuated | 1969 |
| Smallpox vaccine, live, attenuated | 1903 |
| Tetanus and diphtheria toxoids, adsorbed (adult use, Td) | 1955 |
| Tetanus toxoid adsorbed | 1949 |
| Tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis vaccine, absorbed | 2005 |
| Typhoid vaccine (polysaccharide) | 1994 |
| Typhoid vaccine; live, attenuated (oral) | 1990 |
| Varicella vaccine; live, attenuated | 1995 |
| Yellow fever vaccine; live, attenuated | 1953 |
| Zoster vaccine; live, attenuated | 2006 |
* As of December 2012.
TABLE 321-4
Immune Globulin Preparations Made from Human Plasma*
| NAME | ABBREVIATION | ROUTE OF ADMINISTRATION | YEAR LICENSED |
| Immune globulin | IG | Intramuscular | 1943 |
| Hepatitis B immune globulin | HBIG | Intramuscular | 1977 |
| Rabies immune globulin | RIG | Intramuscular | 1974 |
| Tetanus immune globulin | TIG | Intramuscular | 1957 |
| Varicella-zoster immune globulin | VariZIG | Intramuscular | 2012† |
| Immune globulin intravenous | IGIV | Intravenous | 1981 |
| Cytomegalovirus immune globulin intravenous | CMV IGIV | Intravenous | 1990 |
| Botulism intravenous immune globulin | Baby BIG | Intravenous | 2003 |
| Vaccinia immune globulin intravenous | VIG-IGIV | Intravenous | 2005 |
| Immune globulin subcutaneous | IG-SQ | Subcutaneous | 2006 |
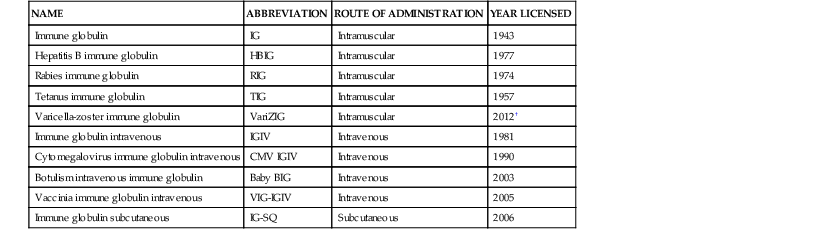
* Antitoxin preparations from animal sera other than humans are available for botulism and diphtheria.
† A previous preparation of varicella-zoster IG (VZIG) was licensed in 1980.
Vaccines
Anthrax Vaccine
Anthrax vaccine (AVA) is prepared from microaerophilic cultures of an avirulent nonencapsulated strain of Bacillus anthracis. The vaccine is a cell-free filtrate that contains a mixture of components, including protective antigen (the antigen that is thought to confer immunity) as well as other bacterial products adsorbed to aluminum hydroxide. Because of concerns about potential use of B. anthracis as a biologic warfare agent, vaccination of selected members of the U.S. Armed Forces was begun in 1998. After the intentional release of anthrax in the United States in 2001, anthrax vaccine was recommended for civilians at risk for repeated exposure to B. anthracis spores, including laboratory personnel handling environmental specimens and performing confirmatory testing for B. anthracis in selected laboratories and workers making repeated entries into sites known to be contaminated with B. anthracis spores. Anthrax vaccine also was used after exposure, in conjunction with antimicrobial prophylaxis, under an investigational protocol.73 Groups for whom preexposure vaccination is recommended include persons working with production quantities of B. anthracis cultures or in activities with a high potential for aerosol production and selected other workers at high risk for exposure to B. anthracis spores.74 Efficacy has been demonstrated in protection against cutaneous disease. Data on clinical efficacy against inhaled anthrax in humans are limited, but available human and animal data are consistent with protection.75 The vaccine induces antibodies in greater than 90% of adults who received the currently recommended primary course of three IM injections given at time zero, 4 weeks, and 6 months, with boosters at 12 months and 18 months, followed by annual boosters.74,76 A controlled study of a vaccine similar to the currently available vaccine demonstrated protective efficacy against cutaneous disease of 93% among mill workers.77 Experience suggests that two doses of vaccine confer some protection.78 Mild local reactions at the site of injection occur in about 30% of recipients. Studies of adverse events after injection of the alum-precipitated vaccine, which is the precursor to the AVA vaccine, demonstrate that more severe local reactions occur infrequently (<4%) and systemic reactions are rare (0.2%). Surveillance for adverse events in the military program revealed no pattern of serious adverse events.79,80 Adverse events, including injection site reaction incidence and duration, were less often seen after IM injection compared with SC injection.76 The IM route of administration is indicated for preexposure use.74 Vaccines containing only recombinant protective antigen are under active development and may be less reactogenic than the current vaccine.81,82 In the event of exposure to anthrax spores, the recommended postexposure prophylaxis regimen is three doses of AVA administered at 0, 2, and 4 weeks, combined with 60 days of antibiotics.74
Calmette-Guérin Bacillus Vaccine
BCG vaccine contains living Calmette-Guérin bacillus, an attenuated strain of Mycobacterium bovis. In many countries, BCG is used in infants and young children to prevent disseminated tuberculosis infection. In the United States, use of BCG is recommended only in special circumstances because the general risk for infection is low. BCG vaccination can also result in conversion of the purified protein derivative (PPD) or Mantoux tuberculin skin test, thereby removing one of the most important indicators of tuberculosis infection (tuberculin conversion). However, the association of a positive PPD skin test after immunization with BCG in childhood tends to fade over time, and most individuals will have a PPD reaction of less than 10 mm by 10 years later. BCG does not cross react with the interferon-gamma release assay (IGRA), so the IGRA is the preferred test over the PPD for diagnosis of tuberculosis in patients older than 4 years who have received BCG.83 The IGRA is not as sensitive in children 4 years or younger and requires a blood draw. Although BCG is widely used throughout the world, there has been much controversy regarding its efficacy. Studies suggest that the vaccine is effective, particularly for preventing complications of disseminated tuberculosis in young children.84–86 In the United States, use of BCG should be considered for individuals, such as infants, whose skin test results are negative and who have prolonged, close contact with patients with active tuberculosis who are untreated, are ineffectively treated, or have antibiotic-resistant infection. BCG also may be considered for health care providers in areas in which multidrug-resistant Mycobacterium tuberculosis infection has become a significant problem.87
A single dose of vaccine is administered intradermally or by the percutaneous route. (The Tice strain licensed in the United States is only approved for percutaneous administration.) Known adverse effects include regional adenitis, disseminated BCG infection, and osteitis caused by the BCG organism. Adenitis occurs in about 1% to 10% of vaccinees, whereas disseminated infections and osteitis are quite rare (about 1 case per 1 million vaccinees). Hypertrophic scars at the injection site occur in up to one third of vaccinated persons, and keloids occur in 2% to 4%. Immunocompromised individuals should not receive the vaccine because of increased risk for disseminated BCG infection.87
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


