Hypoglycemia
Benjamin Glaser
Gil Leibowitz
Hypoglycemia, simply defined as a low blood glucose level, is a laboratory finding that may or may not be associated with significant pathology. Pathologic hypoglycemia may be caused by a large spectrum of entities, which vary depending on the age at onset and the presence of other, related symptoms. In this chapter we will provide a systematic approach to the diagnosis and treatment of hypoglycemic disorders in adults and children.
REGULATION OF GLUCOSE LEVELS (GLUCOSE HOMEOSTASIS)
The central nervous system (CNS) is dependent on the oxidation of glucose as its primary source of energy. Lack of an adequate glucose supply results in brain dysfunction (neuroglycopenia) and, if prolonged, in irreversible neuronal damage and death. In the healthy adult, the glucose requirements of the brain have been estimated at about 1 mg/(kg · min), corresponding to about 100 g/day in a 70-kg man. Uptake of glucose by the CNS is facilitated by two glucose transporters, GLUT1 and GLUT3, neither of which is regulated by insulin. During hypoglycemia, this glucose transport system is rate-limiting. Chronic hypoglycemia is associated with upregulation of glucose transport, a phenomenon that may be important in the development of hypoglycemia unawareness (see below).
In the fasting state, the brain can utilize ketone bodies (β-hydroxybutyrate and acetoacetate) as alternate energy sources. The uptake of ketone bodies by the brain is proportional to their concentration in the blood. Oxidation of ketone bodies can provide a significant energy source only when circulating levels are elevated, as in the case of a prolonged fast. Therefore, when glucose levels are low, but ketone levels are very high, the brain is partially protected from the deleterious effects of hypoglycemia. However, when both glucose and ketone levels are low, as in insulin-induced hypoglycemia and disorders of fatty acid oxidation, the brain is particularly vulnerable to metabolic damage.
Given the importance of glucose for neural function, it is not surprising that redundant mechanisms have evolved to prevent hypoglycemia. The circulating glucose concentration is determined by the balance between glucose input (absorption plus production) and glucose utilization. During fasting, glucose production is dependent on the availability of the substrates and the metabolic machinery required for glycogenolysis and gluconeogenesis. Glucose utilization is determined by insulin-stimulated
glucose uptake as well as by the availability of alternative energy sources, particularly for muscle (1).
glucose uptake as well as by the availability of alternative energy sources, particularly for muscle (1).
The major mechanisms responsible for prevention of hypoglycemia are shown in Figure 69.1. In the postabsorptive state, insulin levels fall, resulting in decreased uptake of glucose by the liver, muscle, and fat and relieving the insulin-mediated suppression of glycogenolysis, gluconeogenesis, and lipolysis. Hepatic glycogenolysis provides most of the glucose requirements during the first 12 to 24 hours of fasting (2), provided that the glycogen stores are adequate and that the uptake of glucose by muscle and fat is not excessive. During more prolonged fasting, after liver glycogen stores are depleted, lipolysis and protein breakdown supply a steady stream of fatty acids, glycerol, and amino acids. The fatty acids are used by muscle as an energy source and by the liver to produce ketone bodies, which can then be used as an energy source by most tissues of the body. Glycerol and amino acids are taken up by the liver and kidney and used as substrates for gluconeogenesis. A recent study demonstrated that net glucose production in healthy males was about 1.8 mg/(kg · min) during a fast lasting up to 40 hours. The contribution of gluconeogenesis to this basal glucose production increased from 41% after 12 hours to 92% after 40 hours of fasting (3). In prolonged fasting, the kidney produces 25% or more of the total glucose requirement, achieved primarily through gluconeogenesis from glutamine, lactate, and glycerol (4). The importance of renal glucose production is emphasized by the finding that severe chronic renal insufficiency may be associated with fasting hypoglycemia (see below).
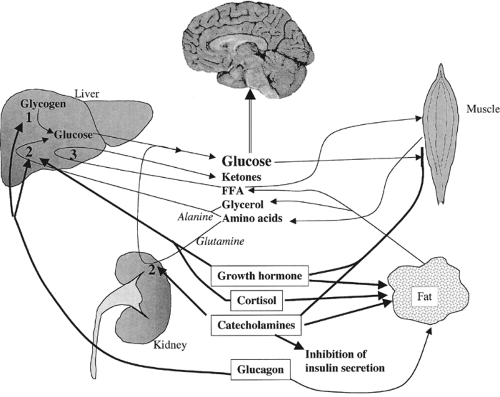 Figure 69.1. Schematic diagram showing the major pathways by which plasma glucose concentrations are maintained in the fasting state. Glucose uptake by the brain is obligatory and constant and is not dependent on hormonal action. During short-term fasting, hepatic glycogen is broken down to glucose by the glycogenolysis pathway (1). During longer fasts, the breakdown of muscle and fat provides the liver with the necessary substrates for gluconeogenesis (2) and ketogenesis (3). Hepatic gluconeogenesis (2) depends primarily on the availability of alanine, whereas renal gluconeogenesis uses glutamine as its primary substrate. Glucagon prevents hypoglycemia, primarily by stimulating hepatic glycogenolysis and gluconeogenesis. Catecholamines stimulate both hepatic and renal gluconeogenesis, while stimulating lipolysis, inhibiting glucose uptake by muscle, and directly inhibiting insulin secretion. Cortisol and growth hormone stimulate both hepatic gluconeogenesis and lipolysis, whereas growth hormone also suppresses peripheral utilization of glucose. |
If glucose levels dip below the threshold of hypoglycemia, counterregulatory hormones are released, resulting in a further increase in glucose production (see below). This threshold has been estimated at 67 mg/dL (i.e., just below the normal glucose level in the postabsorptive state and above the level at which neuroglycopenic symptoms usually occur) (5). The ventromedial hypothalamus appears to be of primary importance in initiating the counterregulatory response (6). The counterregulatory hormones can be divided into two major groups: the rapid-acting hormones (catecholamines and glucagon) and the slow-acting hormones (growth hormone and cortisol).
The catecholamines (epinephrine and norepinephrine) act by further suppressing insulin release while directly stimulating hepatic and renal gluconeogenesis, inhibiting peripheral utilization
of glucose, and stimulating lipolysis. The latter provides both substrate for glyconeogenesis (glycerol) and an alternative energy source for muscle (fatty acids and ketone bodies). Glucagon acts primarily by stimulating hepatic glucose production, having little or no effect on peripheral glucose utilization or renal glucose production. Although glucagon stimulates lipolysis and thus ketogenesis, it appears to have a minimal net effect on the mobilization of gluconeogenic precursors from fat (7). Catecholamines and glucagon act independently, and both are required for an adequate glycemic response. Failure of either will significantly impair the glycemic response to hypoglycemia (8).
of glucose, and stimulating lipolysis. The latter provides both substrate for glyconeogenesis (glycerol) and an alternative energy source for muscle (fatty acids and ketone bodies). Glucagon acts primarily by stimulating hepatic glucose production, having little or no effect on peripheral glucose utilization or renal glucose production. Although glucagon stimulates lipolysis and thus ketogenesis, it appears to have a minimal net effect on the mobilization of gluconeogenic precursors from fat (7). Catecholamines and glucagon act independently, and both are required for an adequate glycemic response. Failure of either will significantly impair the glycemic response to hypoglycemia (8).
The counterregulatory effects of cortisol and growth hormone take several hours to become evident. Therefore, these hormones play a small role in the prevention of acute hypoglycemia but are important in the prevention of hypoglycemia during prolonged fasting. Cortisol stimulates both hepatic gluconeogenesis and lipolysis, resulting in increased levels of free fatty acids and glycerol. Growth hormone has a similar effect on lipolysis and gluconeogenesis, while simultaneously suppressing peripheral utilization of glucose. Both of these hormones rely on increased lipolysis to provide substrates for gluconeogenesis, as well as free fatty acids and ketone bodies, which are used as alternative energy sources. These are in effect glucose sparing in that they decrease the dependence of muscle and other non-neural tissues on glucose. This emphasizes the importance of the lipolytic and ketogenic pathways in the prevention of hypoglycemia. If levels of fatty acids or ketone bodies fail to increase, either because of inadequate fat stores or because of metabolic defects that prevent their production, utilization of glucose by muscle will increase, as will the risk of hypoglycemia, even if the remaining counterregulatory mechanisms function normally.
Therefore, despite the multiple redundant mechanisms that have evolved to prevent hypoglycemia, malfunction of any one of them will place the patient at risk of developing hypoglycemia. The specific nature of the malfunction will determine the associated clinical syndrome, with some defects resulting in profound, treatment-resistant hypoglycemia and others causing only mild hypoglycemia during prolonged fasting. In this chapter we review the common and rare causes of hypoglycemia. Because of the complexity of this problem in the developing child, and because of recent major advances in this field, a separate section is devoted to causes and treatment of hypoglycemia in the newborn and infant.
MEASUREMENT OF CIRCULATING GLUCOSE LEVELS
Accurate measurement of the circulating concentration of glucose is crucial to making the diagnosis of hypoglycemia. Appropriate reference standards must be used, because levels differ depending on whether whole blood or plasma is measured and if the sample is obtained from an artery, vein, or capillary. Most reference ranges are based on venous plasma samples. Arterial glucose concentrations are higher than venous glucose concentrations because of substantial glucose extraction across the forearm during hyperinsulinemic conditions. Glucose concentrations in whole blood are lower than those in plasma because glucose is metabolized by the glycolytic pathway in red blood cells and leukocytes, resulting in unequal distribution of glucose between intracellular and extracellular fluid. Whole-blood glucose measurements may be spuriously high in severe anemia and low in polycythemia vera or in the presence of excessive leukocytosis. Spuriously low glucose measurements due to glycolysis in blood cells in vitro (pseudohypoglycemia) should be avoided by transferring plasma rapidly to the laboratory for centrifugation. If delay is anticipated, chilling on ice will decrease the rate of in vitro glycolysis. Alternatively, this artifact can be minimized by collecting the blood in a tube containing inhibitors of glycolysis, such as oxalate and fluoride.
Glucose values frequently are measured in capillary blood obtained by a fingerstick and applied to a reagent strip for analysis by visual inspection or by a glucose meter. Visual inspection of a reagent strip to which blood has been applied is not a reliable way to distinguish normal from low glucose values. Glucose-meter measurements may be subject to artifacts resulting from poor technique of the patient or the medical staff performing the test or from technical problems with the glucose meter. Moreover, most glucose meters are calibrated to measure high glucose values and thus may be inaccurate in the hypoglycemic range. Therefore, although glucose meters may be very useful for screening patients to determine if symptoms correlate with low glucose measurements, thus excluding hypoglycemia when levels are high, the definitive diagnosis of hypoglycemia must always be based on an appropriately calibrated, standard chemical test in the laboratory.
DEFINITION OF HYPOGLYCEMIA
The diagnosis of hypoglycemia is most convincingly based on the Whipple triad: symptoms consistent with hypoglycemia, a low plasma glucose concentration, and relief of symptoms after the plasma glucose level is raised to normal by administration of exogenous glucose (9). However, the cutoff glucose concentration for defining hypoglycemia is controversial. A review of the literature reveals that glucose concentrations that have been proposed range from 45 to 75 mg/dL (2.5 to 4.2 mmol/L). This controversy stems from the fact that hypoglycemia can be defined in several ways, including the normal response to fasting, the level at which clinical neuroglycopenic symptoms appear, and the level at which a physiologic counterregulation response is triggered. Thus, the glucose level at which hypoglycemia is diagnosed depends primarily on the definition used. According to the first criterion, glucose concentrations in venous plasma obtained after an overnight fast are considered abnormal if they are lower than 60 mg/dL (3.3 mmol/L). However, if hypoglycemia is defined as the clinical pathophysiologic state characterized by autonomic and neuroglycopenic symptoms, then glucose concentrations below 54 mg/dL (3.0 mmol/L) should be defined as pathologic (10).
Defining hypoglycemia is further complicated by the fact that the normal plasma glucose response to prolonged fasting is different in adult men and women. In adult men, the normal fasting plasma glucose level is above 55 mg/dL (3.1 mmol/L) at 24 hours and 50 mg/dL (2.8 mmol/L) at 48 and 72 hours. In normal premenopausal women, fasting plasma glucose levels may dip as low as 35 mg/dL (1.9 mmol/L) at 24 hours, whereas more prolonged fasts may drive glucose levels to as low as 30 mg/dL (1.7 mmol/L) and, rarely, even below this (Fig. 69.2) (11). Responses of counterregulatory hormones to lowered blood glucose are reduced in healthy young women, which may explain these low fasting plasma glucose levels (12). Because the normal glucose response to fasting may vary according to the age and gender of the patient, the appearance of neuroglycopenic symptoms is an important indication that the measurement of a low plasma glucose concentration is indeed true hypoglycemia. In the healthy adult, prolonged fasting results in the formation of ketone bodies that can be used as an alternative fuel for brain metabolism; therefore, neuroglycopenia may not occur at glucose levels that under other conditions would cause severe symptoms. In contrast, hypoglycemia induced by insulin or insulin-like factors is associated with suppression
of ketosis, and the brain may therefore be sensitive to moderate reductions in glucose levels.
of ketosis, and the brain may therefore be sensitive to moderate reductions in glucose levels.
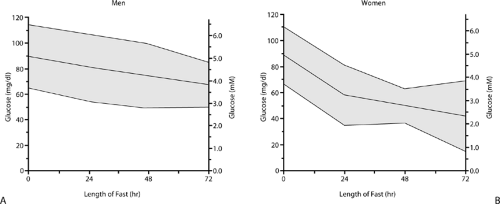 Figure 69.2. Glycemic response to a 72-hour fast in nonobese adult women (A) and men (B). Plasma glucose levels were measured by the glucose oxidase method and are presented as the mean ± 2 standard deviations. (Data from Merimee TJ, Tyson JE. Stabilization of plasma glucose during fasting: normal variations in two separate studies. N Engl J Med 1974;291:1275–1278.) |
An alternative approach is to define hypoglycemia on the basis of the physiologic counterregulatory hormone response to lowering of plasma glucose. Insulin secretion is suppressed at glucose levels of 76 to 72 mg/dL (4.2 to 4.0 mmol/L), and glucose-counterregulatory hormones are released at glucose levels of 70 to 65 mg/dL (3.9 to 3.6 mmol/L) (1).
In practice, the definition of hypoglycemia should be adjusted to the clinical situation. Although there is no well-defined glucose concentration for defining hypoglycemia, venous glucose concentrations between 45 and 60 mg/dL (2.5 and 3.3 mmol/L) strongly suggest hypoglycemia, and those below 45 mg/dL (2.5 mmol/L) are usually indicative of important pathology. If the low glucose level is associated with neurologic symptoms, the clinical suspicion for hypoglycemia is high and prompt evaluation to identify the cause is required. In patients with diabetes treated with insulin, glucose levels should be maintained above 75 mg/dL (4.2 mmol/L) to prevent symptomatic hypoglycemia and hypoglycemia unawareness (see Chapter 40).
SIGNS AND SYMPTOMS OF HYPOGLYCEMIA
Signs and symptoms of hypoglycemia result from neuronal glucose deprivation and can be divided into two categories: autonomic and neuroglycopenic (Table 69.1). The former result from the activation of the autonomic nervous system, with release of epinephrine from the adrenal medulla into the circulation and of norepinephrine from sympathetic postganglionic nerve terminals within the target tissues. Under normal circumstances, the glycemic threshold for catecholamine release is higher than that for induction of neuroglycopenic symptoms. Therefore, the autonomic symptoms precede the neuroglycopenic symptoms. Symptoms and signs related to catecholamine release include tremulousness, pallor, palpitations, tachycardia, widened pulse pressure, and anxiety. Sweating, hunger, and paresthesias are also common and are mediated by acetylcholine. In adults, sweating is often very prominent and is thought to be mediated by sympathetic cholinergic postganglionic neurons (13).
TABLE 69.1. Signs and Symptoms of Hypoglycemia in the Adult | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Neuroglycopenic symptoms are the result of glucose deprivation in the brain. Because glucose is the main fuel of metabolism in brain tissue, reduced levels of glucose may be insufficient to supply the brain with its energy requirements. Thus, the symptoms of neuroglycopenia and those of hypoxia are virtually indistinguishable. They include nonspecific complaints, such as weakness, fatigue and dizziness, headache, behavioral changes, and confusion. Affected persons often show poor judgment, perform their activities in an automatic, repetitive manner, and become uncooperative. They may become lethargic, irritable, and occasionally even aggressive. There is variable impairment of cognitive functions, including difficulty in thinking
and concentration, aphasia, and slurred speech. In addition, hypoglycemia may cause blurred vision, cortical blindness, paresthesias, hemiplegia, hypothermia, and eventually coma, seizures, and death. Symptoms are less specific in the neonate than in the older child and adult and may include irritability, jitteriness, changes in tone, cyanosis, tachypnea, lethargy, and frank coma or seizures (see section on neonatal hypoglycemia).
and concentration, aphasia, and slurred speech. In addition, hypoglycemia may cause blurred vision, cortical blindness, paresthesias, hemiplegia, hypothermia, and eventually coma, seizures, and death. Symptoms are less specific in the neonate than in the older child and adult and may include irritability, jitteriness, changes in tone, cyanosis, tachypnea, lethargy, and frank coma or seizures (see section on neonatal hypoglycemia).
The glycemic threshold for the appearance of counterregulatory hormone response and for neuroglycopenic symptoms is dynamic. It shifts to higher glucose concentrations following sustained hyperglycemia in patients with poorly controlled diabetes (14) and to lower glucose concentrations in patients with recurrent episodes of hypoglycemia (15). Even a single episode of hypoglycemia can blunt catecholamine response to subsequent hypoglycemia (16). The shift in glycemic thresholds following recurrent hypoglycemia results from an adaptive increase in uptake of glucose by the brain, probably due to increased cerebral blood flow (17) and enhanced extraction of glucose induced by upregulation of the brain glucose transporters GLUT1 and GLUT3 (18). In addition to depending on the previous glycemic environment, the response to hypoglycemia depends on the availability of alternative fuels for the brain, such as ketone bodies and lactate (19).
Cerebral energy requirements, the adaptive changes in glucose extraction, and the ability to utilize alternative fuels are not uniform in all the regions of the brain. Thus, enhanced uptake of glucose in response to recurrent hypoglycemia can maintain normal metabolism and function in some regions of the brain, while other regions may remain susceptible to hypoglycemia-induced dysfunction. From the clinical standpoint, a patient with recurrent hypoglycemia may fail to develop warning symptoms of hypoglycemia and may not be able to react quickly enough to avert severe neuroglycopenia and coma when the glucose concentration reaches a critical threshold. Therefore, enhanced uptake of glucose by the brain following recurrent hypoglycemia is potentially dangerous and should be considered maladaptive (18).
Severe and prolonged hypoglycemic episodes may cause neuronal cell death, resulting in permanent impairment of brain function. However, if the hypoglycemia is treated effectively, apparent full recovery is the rule even after severe episodes of hypoglycemia. There are concerns that recurrent hypoglycemia may result in permanent damage to intellectual capacity, although in adults there is no conclusive evidence to support this notion. Prospective studies of patients with diabetes at risk for recurrent severe hypoglycemia show no effect on long-term cognitive function (20,21). In contrast, children younger than 5 years of age do show a permanent impairment in IQ after recurrent events of hypoglycemia (22). Thus, hypoglycemia is harmful to the developing brain during the first years of life.
CLASSIFICATION OF HYPOGLYCEMIC DISORDERS
The differential diagnosis of hypoglycemia is broad (Fig. 69.3). The various hypoglycemic disorders can be classified on the basis of glucose kinetics (increased glucose utilization vs. deficient
glucose production), insulin mediation (hyperinsulinemic vs. hypoinsulinemic), etiology (endogenous vs. exogenous causes), age (adults vs. infants and children), and the clinical characteristics of the patient (appears healthy or ill) (23).
glucose production), insulin mediation (hyperinsulinemic vs. hypoinsulinemic), etiology (endogenous vs. exogenous causes), age (adults vs. infants and children), and the clinical characteristics of the patient (appears healthy or ill) (23).
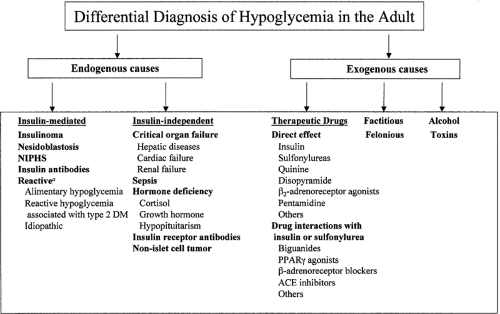 Figure 69.3. The major causes of hypoglycemia in the adult are grouped according to mechanism. NIPHS, noninsulinoma pancreatogenous hypoglycemia; PPAR-γ, peroxisome proliferator-activated receptor-γ ACE, angiotensin-converting enzyme. aPostprandial symptoms are common but usually not associated with hypoglycemia. |
From a clinical standpoint, it is common to classify hypoglycemic disorders as fasting or reactive. Reactive hypoglycemia refers to the occurrence of hypoglycemia in the postprandial state, not during fasting, and usually does not imply a serious underlying disorder. However, this term describes the timing of hypoglycemia and has no implication regarding mechanism or etiology. For example, postprandial hypoglycemia may be the predominant finding in some patients with insulinoma (23). Moreover, the majority of patients referred for evaluation of postprandial symptoms do not have low plasma glucose levels at all. Therefore, true functional/reactive hypoglycemia is not common (24,25).
In this chapter, we refer to hypoglycemic disorders based on the age group of the patients. In adults, hypoglycemia may be caused by drugs, systemic disease, abnormalities in counterregulatory hormones, or tumors. Hypoglycemia arising as a consequence of the therapeutic use of insulin or oral hypoglycemic agents in patients with diabetes, the most common exogenous cause of hypoglycemia, is considered elsewhere in this volume (see Chapters 40 and 41). In infants and nondiabetic children, hypoglycemia often results from inborn genetic defects in proteins important for regulating insulin secretion or substrate metabolism. In recent years, there has been an explosion of information on the human genome, leading to better classification and characterization of different hypoglycemic disorders. Thus, the etiologies of hypoglycemia in this age group are discussed separately.
EXOGENOUS CAUSES OF HYPOGLYCEMIA
Drug-Induced Hypoglycemia
Many drugs have been associated with hypoglycemia (Table 69.2); however, a cause-and-effect relationship has not been established for many of these agents. Factors predisposing to drug-induced hypoglycemia include very young and very old age, impaired renal and liver functions, and poor nutrition (26). Drugs may cause hypoglycemia by several mechanisms: (a) by increasing release of insulin secondary to activation of the insulin secretion machinery or secondary to a direct toxic effect to pancreatic islets resulting in nonregulated insulin release; (b) by increasing uptake and utilization of glucose in the peripheral tissue; and (c) by decreasing production of glucose by the liver. In addition, drugs may interact with sulfonylurea agents and insulin and potentiate their hypoglycemic effects. In these cases it may be difficult to assess the role of the individual drugs in the development of hypoglycemia.
TABLE 69.2. Hypoglycemia-Causing Drugs | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Many of the drugs that have been associated with hypoglycemia are in very common use, and patients, particularly the elderly, are frequently receiving multiple drugs. Therefore, iatrogenic hypoglycemia should always be included in the differential diagnosis. Doing so may facilitate diagnosis and treatment and prevent unnecessary diagnostic and therapeutic procedures. The most common causes of drug-induced hypoglycemia are alcohol and treatment with a sulfonylurea or insulin in patients with diabetes. The latter is discussed in Chapter 41.
DRUGS AND TOXINS THAT INCREASE INSULIN RELEASE
Quinine and Related Drugs
The antimalarial drug quinine and its antiarrhythmic derivative quinidine have been shown to induce hyperinsulinemic hypoglycemia. Quinine-related substances stimulate insulin secretion from pancreatic β-cells and cause a mild decrease in blood glucose levels that is usually not sufficient to induce symptoms of hypoglycemia in healthy subjects. The hypoglycemic effect of quinine may become more prominent in the presence of renal failure, since drug clearance is reduced and the concentration of quinine in the blood is higher. In addition to quinine-induced hyperinsulinemia, acutely ill patients with malaria are prone to development of hypoglycemia because of consumption of glucose by the malarial parasites. In patients with malaria falciparum
who are treated with quinine, hypoglycemia may be overlooked because the neurologic symptoms may be attributed to cerebral malaria. Therefore, blood glucose should be carefully monitored and a glucose supplement administered to prevent dangerous hypoglycemia (27,28).
who are treated with quinine, hypoglycemia may be overlooked because the neurologic symptoms may be attributed to cerebral malaria. Therefore, blood glucose should be carefully monitored and a glucose supplement administered to prevent dangerous hypoglycemia (27,28).
Disopyramide
Disopyramide, used clinically as an antiarrhythmic agent, has been shown to increase insulin secretion from pancreatic β-cells, resulting in hyperinsulinemic hypoglycemia. The drug causes closure of the β-cell KATP channel, binding to the channel at a site distinct from the sulfonylurea binding site (29). Disopyramide often is used in elderly patients, who may also suffer from impaired renal function and who appear to be particularly susceptible to the glucose-lowering effect of the drug (30). A recent study found that the use of disopyramide did not affect the risk of hypoglycemia in an ambulatory patient population, suggesting that this side effect may be minor in the general population and may be of clinical importance only in selected patients at particular risk (31). It has been claimed that, although rare in absolute terms, disopyramide is one of the most common causes of drug-induced hypoglycemia in nondiabetic patients (32).
β2-Adrenoreceptor Agonists
Ritodrine therapy to delay premature delivery during pregnancy can stimulate insulin secretion and cause hypoglycemia in the newborn and rarely in the mother (33). Therapeutic doses of β2-adrenoreceptor agonists administered to patients with bronchial asthma were not reported to induce hypoglycemia; however, albuterol overdose was reported to cause hypoglycemia in a young child (34).
Pentamidine
Pentamidine is used for the treatment of Pneumocystis carinii pneumonia in patients with AIDS and for parasitic infections, including trypanosomiasis and leishmaniasis. The drug is toxic to pancreatic β-cells, resulting in unregulated release of insulin from the damaged β-cells and leading to hypoglycemia followed by hyperglycemia and even permanent diabetes mellitus. The toxicity to the β-cells is more common and severe following systemic administration and is related to the cumulative dose of the drug (35,36).
DRUGS THAT INCREASE INSULIN SENSITIVITY
Biguanides and Peroxisome Proliferator-Activated Recepto-γ Agonists
Metformin and the glitazones do not usually cause hypoglycemia when taken alone. Over 10 years of follow-up among patients with type 2 diabetes taking metformin in the UK Prospective Diabetes Study (UKPDS), the proportion of patients per year who had one or more episodes of hypoglycemia was 4.2%, none of whom developed severe hypoglycemia (37). The biguanides and glitazones may augment hypoglycemia caused by sulfonylureas and insulin by increasing the uptake and utilization of glucose (38). Metformin also decreases hepatic production of glucose and decreases intestinal absorption of glucose.
Angiotensin-Converting Enzyme Inhibitors
The association between angiotensin-converting enzyme (ACE) inhibitors and hypoglycemia in patients with diabetes treated with sulfonylureas or insulin is controversial. One study reported that 14% of all hospital admissions for hypoglycemia might be attributable to ACE inhibitors (39). The association between ACE inhibitors and hypoglycemia was not explained by potential confounding factors, such as the type of diabetes treatment and congestive heart failure (40). ACE inhibitors can intensify the glucose-lowering effect of insulin or sulfonylureas. A study using a hyperinsulinemic euglycemic clamp protocol found that captopril improved sensitivity to insulin in healthy volunteers with normal blood pressure. With enalapril, however, the increase in insulin sensitivity was less pronounced and did not achieve statistical significance (41,42). ACE inhibitors may indirectly increase insulin sensitivity by increasing circulating kinins, leading to vasodilatation and increased uptake of glucose in muscle tissue. Nevertheless, large series of patients with diabetes treated with ACE inhibitors, including the UKPDS, show that these are safe and that the risk of hypoglycemia is very low (43).
β-Adrenergic Receptor Blocking Agents
Nonselective β-adrenergic receptor blocking agents, such as propranolol, have been associated with hypoglycemia in otherwise healthy children but not in healthy adults (44). Most of the cases of hypoglycemia associated with these agents were in patients with diabetes receiving insulin or sulfonylureas. Thus, it is difficult to prove a cause-and-effect relationship between therapy with β-blockers and hypoglycemia (45). β-Blockers can increase glucose uptake in skeletal muscle by antagonizing the effects of catecholamines on glucose uptake and lipolysis (46). Suppression of lipolysis with reduction of nonesterified free fatty acids in plasma improves insulin sensitivity and indirectly reduces gluconeogenesis. β-Blockers decrease insulin secretion from pancreatic β-cells. Therefore, insulin-treated patients with diabetes may be more susceptible to the glucose-lowering effects of β-blockers, whereas patients with type 2 diabetes treated with oral medications or diet may have exacerbations of hyperglycemia rather than hypoglycemia. Similar to ACE inhibitors, β-blockers were not associated with a higher frequency of hypoglycemic episodes in large series of patients with type 2 diabetes, including the UKPDS (43). β-Blockers may block the adrenergic response to hypoglycemia, leading to hypoglycemia unawareness and a predominance of the neuroglycopenic symptoms. Moreover, β-blockers may delay the recovery from hypoglycemia because of inhibition of catecholamine-mediated glucose counterregulation. Catecholamines are not required for prompt recovery from hypoglycemia in patients with intact islet-cell function; however, β-blockade may become critical in glucagon-deficient patients with type 1 diabetes. The risk for severe hypoglycemia in insulin-treated patients with diabetes can be reduced by using one of the relatively selective β1-adrenergic antagonists (47).
DRUGS THAT DECREASE HEPATIC GLUCOSE PRODUCTION
Hypoglycemia resulting from suppressed hepatic production of glucose may occur in the setting of acute drug intoxication, which may result in metabolic inhibition of gluconeogenesis, such as with alcohol or in hepatic necrosis, as may occur after paracetamol poisoning. Hypoglycemia may be exacerbated if acute renal failure occurs concomitantly with hepatic toxicity.
Alcohol-Induced Hypoglycemia
The most common cause of drug-induced hypoglycemia secondary to suppressed gluconeogenesis is alcohol overdose. Alcohol-induced hypoglycemia may occur in persons of all ages and in all classes of society. It is more common in men than in women and in malnourished persons on a low-calorie diet. Young children are especially susceptible to the hypoglycemic effects of alcohol. Therefore, accidental consumption of even modest amounts of alcohol may result in hypoglycemia in infants and young children (48). Fewer than 1% of patients with
alcohol intoxication presenting to the emergency room have hypoglycemia. However, because of the very high incidence of alcohol intoxication relative to the incidence of all other causes of hypoglycemia, alcohol can be implicated in 18% to 52% of patients admitted to the emergency department because of sustained hypoglycemia (26). This variation in the incidence of alcohol-induced hypoglycemia in different series probably reflects social, ethnic, and cultural differences in alcohol consumption among the populations studied. Symptoms of alcohol intoxication may be very similar to those of neuroglycopenia, making clinical differentiation difficult. Thus, immediate routine determination of blood glucose is recommended for all intoxicated patients in the emergency department to guarantee rapid identification of those with significant hypoglycemia. A delay in the diagnosis may result in permanent brain damage and death. The mortality rate among hospitalized patients with alcohol-induced hypoglycemia may be as high as 10% (26).
alcohol intoxication presenting to the emergency room have hypoglycemia. However, because of the very high incidence of alcohol intoxication relative to the incidence of all other causes of hypoglycemia, alcohol can be implicated in 18% to 52% of patients admitted to the emergency department because of sustained hypoglycemia (26). This variation in the incidence of alcohol-induced hypoglycemia in different series probably reflects social, ethnic, and cultural differences in alcohol consumption among the populations studied. Symptoms of alcohol intoxication may be very similar to those of neuroglycopenia, making clinical differentiation difficult. Thus, immediate routine determination of blood glucose is recommended for all intoxicated patients in the emergency department to guarantee rapid identification of those with significant hypoglycemia. A delay in the diagnosis may result in permanent brain damage and death. The mortality rate among hospitalized patients with alcohol-induced hypoglycemia may be as high as 10% (26).
Ethanol is usually still detectable in the blood at the time the patient presents with hypoglycemia but may not be markedly elevated and may correlate poorly with the glucose concentration. Characteristically, the patient with alcohol-induced hypoglycemia is an alcoholic with a history of moderate to excessive consumption of alcohol and little or no protein or carbohydrate intake during the preceding hours or days. The patient often is referred to the hospital in a comatose state and also may suffer from hypothermia. Most patients do not experience autonomic response to alcohol-induced hypoglycemia, and some chronic alcoholics can tolerate low blood glucose levels without symptomatic neuroglycopenia. The diminished neuronal response to hypoglycemia may result from recurrent subclinical episodes of hypoglycemia that reduce the glycemic threshold for the appearance of counterregulatory hormone response and for neuroglycopenic symptoms. There may also be a direct effect of alcohol on the activation of the hypothalamic-adrenal axis in response to hypoglycemia. In addition, plasma ketone bodies are often high and can be used as an alternative fuel for brain metabolism. Some patients present with alcoholic ketoacidosis, which may occur several days following alcohol ingestion and resembles diabetic ketoacidosis. In these patients, plasma glucose levels are usually normal or high; however, some patients may present with severe hypoglycemia. Both the pathogenesis of this condition and the reason certain patients develop hypoglycemia while others become hyperglycemic are unknown. This medical emergency should be treated with intravenous glucose injection preceded by thiamine injection to prevent the development of Wernicke encephalopathy.
Alcohol causes hypoglycemia by inhibiting gluconeogenesis. Ethanol is metabolized in the liver by alcohol dehydrogenase to acetaldehyde and then to acetic acid by aldehyde dehydrogenase. The enzymatic reactions generate free hydrogen ions, with subsequent reduction of nicotinamide dinucleotide (NAD) to NADH. The depletion of hepatic NAD stores inhibits the metabolism of precursors in the gluconeogenic pathway, including the conversion of lactate to pyruvate, several steps in the tricarboxylic acid cycle, and the conversion of glycerol to glucose (49). Ethanol does not inhibit glycogenolysis. Thus, well-nourished obese patients and those who have had significant carbohydrate intake within 12 to 24 hours are likely to have adequate hepatic glycogen stores, which partially protect them from hypoglycemia.
Ethanol also inhibits the release of corticotropin, cortisol, and growth hormone in response to hypoglycemia. The glucagon and epinephrine responses to hypoglycemia may also be delayed, although this is controversial. The failure to activate a counterregulatory hormone response may contribute to the development of fasting hypoglycemia (50).
In addition to inducing fasting hypoglycemia, alcohol may increase the risk for reactive hypoglycemia. Moderate alcohol consumption was shown to increase insulin secretion in response to glucose stimulation. Thus, a mixture of alcohol and sucrose may induce postprandial reactive hypoglycemia due to insulin excess. For example, 10% to 20% of healthy volunteers who consumed a gin and tonic (a sucrose-rich alcoholic beverage) on an empty stomach and did not eat for several hours afterwards developed symptomatic reactive hypoglycemia (51).
Interaction Between Alcohol and Antidiabetic Drugs
The effect of alcohol on insulin-induced hypoglycemia is controversial. In one study, blood glucose levels were similar after administration of insulin and ethanol together or insulin alone (52). However, alcohol does inhibit gluconeogenesis (see above) and has been reported to be commonly involved in admissions to the hospital for hypoglycemia resulting from insulin overdose (53). Furthermore, there are concerns that alcohol consumption may exacerbate the brain-damaging effects of hypoglycemia. There is very limited information on the interaction between sulfonylureas and the hypoglycemic effects of alcohol. In one large series of patients with drug-induced hypoglycemia, a few patients with diabetes developed hypoglycemia following administration of both sulfonylureas and alcohol (54).
Hypoglycin A
Hypoglycemia may occur after the ingestion of the unripe akee fruit. The akee fruit is commonly consumed in Jamaica, and if eaten unripe may cause severe weakness, vomiting, convulsions, coma, and eventually death. The symptoms are associated with severe hypoglycemia resulting from the toxin hypoglycin A, which inhibits fatty acid oxidation and thus causes a metabolic syndrome very similar to that caused by inborn errors of fatty acid metabolism (see section on hypoglycemia in the neonate) (55). Both ketogenesis and hepatic gluconeogenesis are inhibited, causing both hypoglycemia and lowered ketone bodies, a combination that may be particularly neurotoxic.
OTHER DRUGS
Many drugs cause hypoglycemia by various mechanisms, some of which have not yet been completely defined. Some drugs, such as sulfonamide, dicumarol, and phenylbutazone, interact with sulfonylureas to enhance their hypoglycemic activity. Other drugs, including isoniazid and synthetic antimalarial drugs such as chloroquine, may rarely induce hypoglycemia in patients with diabetes by reducing the clearance of insulin from the blood. A large dose of salicylates may induce hypoglycemia in young children, but rarely in adults. The mechanism of salicylate-induced hypoglycemia is unknown but may be related to increased peripheral utilization of glucose, suppression of hepatic gluconeogenesis, or augmentation of insulin release (26). Recent studies have shown that salicylates can enhance insulin sensitivity by inhibiting the IκB kinase (56).
Factitious Hypoglycemia
Most cases of factitious hypoglycemia are caused by the deliberate administration of insulin or oral hypoglycemic agents by the patient, although accidental administration or administration by a caregiver may occur as well. The prevalence of factitious hypoglycemia is unknown. It was suggested that it is more common in women than in men, although this has not been confirmed (57). The typical patient is young, aged 15 to 40 years, and has access to insulin or oral hypoglycemic agents such as sulfonylureas. The patient may be a medical or paramedical
worker or a family member of a person with diabetes treated with oral hypoglycemic agents or insulin and may know the effects of the abused medication and the symptoms of hypoglycemia. Most patients seem to be “mentally healthy” and provide detailed description of their “illness,” obviously omitting the fact that hypoglycemic drugs were abused. Similar to other factitious diseases, use of hypoglycemic drugs is usually denied. When confronted with this possibility, the patient may respond in an aggressive manner, blaming the physician for not trusting him or her. These patients may be very sophisticated in concealing self-administration of hypoglycemic drugs, and episodes of hypoglycemia may occur even during close supervision in the hospital. The history of episodes of hypoglycemia may extend over periods of many years, sometimes with intervals of remission explained by irregular administration of the drugs. The history may suggest the presence of an insulinoma, and failure to diagnose the cause of hyperinsulinism may result in an unnecessary laparotomy and even pancreatectomy.
worker or a family member of a person with diabetes treated with oral hypoglycemic agents or insulin and may know the effects of the abused medication and the symptoms of hypoglycemia. Most patients seem to be “mentally healthy” and provide detailed description of their “illness,” obviously omitting the fact that hypoglycemic drugs were abused. Similar to other factitious diseases, use of hypoglycemic drugs is usually denied. When confronted with this possibility, the patient may respond in an aggressive manner, blaming the physician for not trusting him or her. These patients may be very sophisticated in concealing self-administration of hypoglycemic drugs, and episodes of hypoglycemia may occur even during close supervision in the hospital. The history of episodes of hypoglycemia may extend over periods of many years, sometimes with intervals of remission explained by irregular administration of the drugs. The history may suggest the presence of an insulinoma, and failure to diagnose the cause of hyperinsulinism may result in an unnecessary laparotomy and even pancreatectomy.
Accidental administration of insulin or oral hypoglycemic agents may produce “factitious” hypoglycemia. Inadvertent administration of insulin to the wrong patient in a hospital is a rare cause of hypoglycemia. Unintended use of oral hypoglycemic agents may result from a prescribing error by a physician or a pharmacist or by substitution of an oral hypoglycemic for another medication by the patient or by a family member. Insulin and drug-induced factitious hypoglycemia may also be the result of attempted suicide or homicide.
The diagnosis of factitious hypoglycemia should be considered in all patients with hyperinsulinemic hypoglycemia. The initial history may provide clues suggesting the presence or absence of factitious hypoglycemia; however, each patient must be fully evaluated regardless of initial impression. We recently evaluated a young soldier with a highly suggestive history of insulinoma, no apparent access to insulin or drugs, and no suggestion of secondary gain, who was subsequently proven to have ingested glibenclamide. Another patient, with a clinical history highly suspicious of factitious hypoglycemia and easy access to medical personnel and medication, was subsequently proven to have a benign insulinoma.
The biochemical diagnosis of insulin-induced factitious hypoglycemia is relatively simple if blood is collected during an episode of hypoglycemia for glucose, insulin, and C-peptide measurement. Plasma insulin is high, with discordant low levels of C-peptide, reflecting the suppression of endogenous insulin secretion by low glucose levels. If insulin and C-peptide measurements are discordant, the presence of antibodies to insulin should be excluded. Insulin antibodies may cause spuriously high or low insulin measurements, depending on the method used in the insulin assay. The presence of circulating antibodies should also be excluded by direct measurement using specific assays and by measuring insulin levels on serial dilutions of plasma samples. Nonlinear correlation between measured insulin and plasma dilution indicates the presence of a substance that interferes with the insulin assay. In the past, the presence of insulin antibodies was thought to be prima facie evidence of exogenous insulin administration. However, modern, highly purified, recombinant human insulin rarely causes clinically significant antibody production, and it is now clear that antibodies may also occur in rare patients with autoimmune hypoglycemia and prior to the clinical onset of type 1 diabetes. Therefore, detection of insulin antibodies should not be regarded as a specific and sensitive assay for the diagnosis of factitious hypoglycemia. In the past, all commercially available insulin was obtained from beef or porcine pancreas. Thus, species-specific assays could be developed to prove the exogenous origin of circulating insulin. Today, most commercial insulin is of human origin, making these assays obsolete; however, the increasing use of insulin analogues may cause a revival of this approach.
In contrast to surreptitious administration of insulin, secretagogue-induced hypoglycemia is associated with elevation of both insulin and C-peptide levels. Plasma levels of C-peptide fail to be suppressed in response to the insulin hypoglycemia test, and the response to different stimuli, such as glucagon and glucose, may be excessive, similar to the findings in patients with insulinoma. Therefore, the diagnosis of factitious hypoglycemia due to insulin secretagogues is usually a difficult challenge. For confirmation or exclusion of the diagnosis, blood levels of some of the relevant drugs should be measured during hypoglycemia in an experienced toxicology laboratory. The increasing number of drugs available that stimulate insulin secretion is making this task progressively more difficult.
On certain occasions, when there is strong clinical suspicion of factitious hypoglycemia, it may be necessary to search the hospital room of a patient to find insulin vials, syringes, needles, or drug tablets to confirm the diagnosis of factitious hypoglycemia. A room search is considered by some to be unethical and should be performed only after careful consideration and approval of the appropriate hospital authorities.
The treatment of factitious hypoglycemia is the same whether it is mediated by insulin or sulfonylurea and includes immediate relief of hypoglycemia by intravenous glucose followed by psychiatric intervention to prevent recurrent episodes of hypoglycemia. Management involves a supportive confrontation followed by long-term psychiatric care. Factitious hypoglycemia is a serious psychiatric disturbance. Although some patients admit that the motive is to gain attention, sympathy, or some other secondary gain, in the majority insight is limited and the long-term prognosis is poor. Hypoglycemia may recur or may be followed by other psychosomatic symptoms or drug abuse. Some patients may later commit suicide (58).
ENDOGENOUS CAUSES OF HYPOGLYCEMIA
Hypoglycemia Due to Critical-Organ Failure
Sepsis and diseases that cause severe hepatic, cardiac, or renal failure are common causes of hypoglycemia among hospitalized patients. Patients with malnutrition or with failure of more than one critical organ are more susceptible to severe hypoglycemia.
HEPATIC DISEASES
Hepatic glycogenolysis and gluconeogenesis play critical roles in the maintenance of postabsorptive blood glucose. Since the normal liver has a very large capacity for glucose production, hypoglycemia will occur only in the face of massive destruction of the liver parenchyma or in the presence of accelerated utilization of glucose. Accelerated utilization of glucose may be present in several clinical conditions, including treatment with insulin or oral medications and during sepsis. Hypoglycemia has been reported in patients with severe liver failure due to toxic hepatitis, fulminant viral hepatitis, fatty liver, or hepatitis associated with alcohol administration and cholangitis. In the latter, sepsis complicating biliary obstruction may contribute to the development of hypoglycemia. Hypoglycemia is uncommon in patients with cirrhosis. Plasma levels of insulin may be relatively high because of the presence of portal-systemic shunts and reduced hepatic insulin extraction (59).
CARDIAC FAILURE
Occasionally, hypoglycemia may occur in patients with severe congestive heart failure. The primary mechanism causing hypoglycemia in these patients is not known but may be the combination of cachexia, lack of gluconeogenic substrate, and liver dysfunction due to hepatic hypoxia and congestion. Patients admitted to intensive care units may develop ischemic hepatitis resulting from episodes of acute heart failure and hypotension. A retrospective study of patients with ischemic hepatitis showed that hypoglycemia is common, occurring in one third of the patients (60).
RENAL FAILURE
Renal insufficiency may be associated with hypoglycemia, particularly in patients with end-stage renal disease (61). In one retrospective study, 3.6% of patients with end-stage renal failure admitted to the hospital had hypoglycemia (62). The most common associated pathologies were drug-induced hypoglycemia and sepsis. Severe malnutrition was associated with 7% of hypoglycemic episodes in these patients. Hypoglycemia associated with either sepsis or malnutrition was associated with a particularly high mortality.
Diabetic and nondiabetic patients undergoing either hemodialysis or peritoneal dialysis can develop spontaneous hypoglycemia during or immediately after dialysis (63). This is thought to be caused by increased insulin secretion induced by a high glucose concentration in the dialysis fluid combined with reduced renal insulin clearance. In addition, hypoglycemia may occur when the patients are dialyzed with a glucose-free dialysis fluid. Patients with an initially low plasma glucose level and those who do not eat during dialysis are particularly at risk for hypoglycemia. The hormonal response to hypoglycemia is blunted, and hypoglycemia unawareness is common (64).
The pathogenesis of hypoglycemia in renal failure involves multiple mechanisms, including reduced supply of substrate for gluconeogenesis, especially in patients with severe cachexia, malnutrition with decreased carbohydrate intake, resistance to the hyperglycemic effect of glucagon, and inhibition of gluconeogenesis (65). In healthy volunteers, renal glucose production accounts for 25% of systemic glucose appearance in the postabsorptive state. Furthermore, epinephrine infusion, which increased plasma epinephrine to levels observed during hypoglycemia, increased the renal release of glucose nearly twofold so that, at the end of the infusion, renal glucose release accounted for 40% of systemic glucose production and essentially all of the increase due to adrenergic stimulation (66). Thus, renal glucose production may have an important role in glucose homeostasis in the postabsorptive state and during hypoglycemia. Patients with renal failure are therefore more susceptible to hypoglycemia (67). A considerable number of patients with end-stage renal failure have diabetes. These patients may develop hypoglycemia as a result of decreased clearance of hypoglycemic drugs or insulin.
Sepsis
Sepsis frequently is associated with hypoglycemia in hospitalized patients. In experimental animals, there are several metabolically distinct phases. In the early phase of sepsis, glucose production and utilization are increased and the animals may be hyperglycemic. Hyperglycemia is followed by a hypoglycemic phase in which hepatic glucose production is decreased. This phase is characterized by a rapid depletion of hepatic glycogen content, impaired glycogenesis, and depressed gluconeogenesis, combined with increased peripheral utilization of glucose (68). Regulation of glucose metabolism by the liver is a complicated process that includes several hormonal regulatory factors, such as insulin, glucagon, and catecholamines. Transcriptional and posttranscriptional regulation of β2-adrenergic receptor gene expression may be responsible for the altered hepatic glucose metabolism during the progression of sepsis (69). Increased utilization of glucose during sepsis is explained by increased use of glucose by macrophage-rich tissues such as liver, spleen, and lung (70). The muscle and fat cells make only a only modest contribution to the increase in glucose utilization. Cytokines such as interleukin-1, interleukin-6, and tumor necrosis factor-α released during sepsis can increase insulin secretion and stimulate glucose transport, leading to imbalance between glucose production and utilization (71,72). Some of these factors have also been implicated in induction of insulin resistance in states of sepsis.
Endocrine Deficiency Disorders and Hypoglycemia
Decreased secretion of multiple glucoregulatory hormones secondary to recurrent hypoglycemia in patients with insulinoma or type 1 diabetes may exacerbate hypoglycemia and prevent prompt recovery of glucose levels. However, in the adult, hormone deficiencies alone are rarely directly responsible for spontaneous hypoglycemia, since insulin secretion is suppressed in response to hypoglycemia and the counterregulatory mechanisms are partially redundant.
Glucagon and Catecholamine Deficiency
Glucagon and epinephrine are important in the counterregulatory response to short-term hypoglycemia. Glucagon increases endogenous glucose production by stimulating hepatic glycogenolysis and gluconeogenesis. In contrast to glucagon, which exclusively affects glucose production, catecholamines have multiple effects on glucose homeostasis, including suppression of endogenous insulin secretion, stimulation of gluconeogenesis and glycogenolysis, and inhibition of glucose utilization. The relative importance of glucagon compared with catecholamines in the physiologic response to hypoglycemia is controversial; however, deficiency of both glucagon and epinephrine in the presence of insulin may result in severe hypoglycemia (73). In clinical practice, isolated deficiency of glucagon and/or epinephrine in patients without diabetes is extremely rare. There are very few case reports of children with hypoglycemia that was presumed to be due to selective deficiency of epinephrine or glucagon (74,75). In some patients with presumed glucagon or epinephrine deficiency, other causes of hypoglycemia, such as hyperinsulinism, were not excluded and the secretion of other glucoregulatory hormones was not studied. Hypoglycemia does not occur in patients receiving glucocorticoid and mineralocorticoid replacement therapy after bilateral adrenalectomy and in patients treated with adrenoreceptor blockers (73). Thus, isolated deficiencies of epinephrine or glucagon rarely, if ever, cause spontaneous hypoglycemia in the absence of hyperinsulinism or an additional factor that decreases glucose levels.
GROWTH-HORMONE AND CORTISOL DEFICIENCIES
In contrast to the rapid-acting hormones glucagon and epinephrine, growth hormone and cortisol are more important in the late response to hypoglycemia. The counterregulatory effects of these hormones are evident after more than 3 hours of hypoglycemia. Growth hormone and cortisol increase plasma levels of free fatty acids and glycerol, resulting in suppression of glucose utilization and an increase in gluconeogenesis. In addition, cortisol increases gluconeogenesis by induction of
hepatic gluconeogenic enzymes (1). The major glucoregulatory effects of growth hormone and cortisol are on hepatic gluconeogenesis. Therefore, deficient growth hormone and/or cortisol may cause hypoglycemia, especially after a prolonged fast when hepatic glycogen stores are depleted. The problem may be compounded when both hormones are deficient, as seen in patients with hypopituitarism.
hepatic gluconeogenic enzymes (1). The major glucoregulatory effects of growth hormone and cortisol are on hepatic gluconeogenesis. Therefore, deficient growth hormone and/or cortisol may cause hypoglycemia, especially after a prolonged fast when hepatic glycogen stores are depleted. The problem may be compounded when both hormones are deficient, as seen in patients with hypopituitarism.
In clinical practice, however, most adults with growth-hormone and/or cortisol deficiency do not develop spontaneous hypoglycemia. Fasting plasma glucose concentrations of patients with primary adrenal insufficiency and patients with corticotropin deficiency are similar to those in normal controls (76). Plasma glucose concentrations after prolonged insulin infusion were lower in patients with panhypopituitarism; however, the recovery from hypoglycemia was intact (77), and the risk of hypoglycemia is not higher compared with that in patients with isolated deficiency of growth hormone or corticotropin. When present, the tendency to develop hypoglycemia following prolonged fasting can be corrected by glucocorticoid replacement, whereas growth-hormone replacement has minimal effect on fasting glucose (77). Adults with hypopituitarism may develop hypoglycemia when glucose production is decreased, as during alcohol ingestion or sepsis, or when glucose utilization is increased, such as during vigorous exercise or pregnancy. Newborns and young children with hypopituitarism are more susceptible to hypoglycemia. Thus, cortisol and growth hormone are probably more important in glucose counterregulation in early life. This hypothesis is supported by experimental data showing that glucocorticoid-deficient newborn mice develop hypoglycemia more frequently during fasting than do adult mice (78).
Immune Hypoglycemia
Hypoglycemia may rarely result from autoantibodies directed against insulin or the insulin receptor. The antibodies disrupt the synchronous coupling between changes in blood glucose and insulin action, thus leading to hypoglycemia.
HYPOGLYCEMIA CAUSED BY ANTIBODIES TO INSULIN
The most common cause for the presence of circulating insulin antibodies is the administration of insulin to patients with diabetes. The antibodies may reduce the levels of free insulin postinjection, resulting in higher postprandial levels of glucose, but may also increase the half-life of insulin (79). Theoretically, a prolonged half-life of insulin causes late hypoglycemia after a bolus injection of insulin. In practice, however, the antibodies do not have a major effect on glycemic control and the incidence of severe hypoglycemia (80). Antibodies to insulin also may develop in patients with recent-onset type 1 diabetes before insulin treatment, relatives of patients with type 1 diabetes or other autoimmune disorders, and occasionally in otherwise healthy subjects (81).
In rare patients, insulin antibodies may be a primary cause of hypoglycemia. The majority of patients with hypoglycemia induced by antibodies to insulin are Japanese. The higher prevalence of autoimmune hypoglycemia in the Japanese population is probably explained by the higher prevalence of specific HLA class 2 alleles that are strongly associated with the disease in this population (82). Hypoglycemia is often postprandial, although fasting hypoglycemia may also occur. Patients often develop hyperglycemia immediately after a meal or glucose load followed by hypoglycemia 2 to 3 hours later. The severity of hypoglycemia varies, and the presentation may be of severe neuroglycopenia with symptoms of confusion, cognitive dysfunction, and even coma. Patients may have other autoimmune diseases, such as Graves disease, systemic lupus erythematosus, or rheumatoid arthritis (80). Exposure to sulfhydryl-containing medications, especially methimazole, was reported for almost half of the patients (82). Interferon-α and medications that cause drug-induced lupus, such as hydralazine, procainamide, and isoniazid, also may cause the development of insulin antibodies and hypoglycemia. Several patients with plasma-cell dyscrasias were reported to have hypoglycemia due to monoclonal insulin-binding antibodies with a low affinity for insulin and a high binding capacity (80). Insulin levels in patients with insulin antibodies causing hypoglycemia usually are high (>100 μU/mL). The endogenous insulin antibodies may interfere with the insulin assay, leading to spurious high or low results, depending on the insulin assay used. C-peptide levels are not fully suppressed because insulin is endogenously secreted. In contrast, surreptitious insulin injection leads to high plasma insulin levels together with complete suppression of C-peptide secretion. The presence of insulin antibodies can be confirmed by high binding of radioactively labeled insulin to polyethylene glycol-precipitated serum samples or by enzyme-linked immunosorbent assay (81). Insulin autoantibodies associated with hypoglycemia cannot be distinguished from those found in insulin-treated patients with diabetes.
The pathogenesis of hypoglycemia caused by insulin antibodies reflects the equilibrium between bound and free insulin, leading to inappropriate release of free insulin that dissociates from the antibodies while the blood glucose concentration is already decreased. Other possible mechanisms, including enhancement of insulin binding to its receptor resulting from cross-linking by the insulin antibodies or direct stimulation of insulin secretion by insulin antibodies, may be operative in some patients (80).
Most patients with autoimmune hypoglycemia due to insulin antibodies experience spontaneous remission (82). If autoimmune hypoglycemia is precipitated by a medication, it should be discontinued and the problem of hypoglycemia will resolve. Patients with recurrent nonremitting hypoglycemia may benefit from frequent low-carbohydrate meals to reduce postprandial hyperglycemia and insulin secretion. Medications that reduce glucose absorption or insulin secretion, such as acarbose, diazoxide, and octreotide, have also been used; however, the results are not consistent. Finally, glucocorticoid therapy and plasmapheresis have also been tried, with the goal of lowering the titer of insulin antibodies (80).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


