Pathogenesis of Cardiovascular Disease in Diabetes
Edward P. Feener
Victor J. Dzau
Diabetes mellitus is a major independent risk factor for cardiovascular disease (CVD) (1). The increased prevalence of CVD in diabetes has been attributed in large part to the acceleration of coronary atherosclerosis, which occurs at an earlier age and advances more rapidly to clinical cardiovascular events in individuals with diabetes than in those without diabetes (2,3). Patients with diabetes are also prone to arterial thrombosis due to persistently activated thrombogenic pathways and impaired fibrinolysis (4,5,6,7). This combination of increased arterial disease and prothrombotic milieu in diabetes is a major underlying cause of acute ischemic coronary heart disease (CHD). Moreover, CHD in diabetes is often diffuse, with an increase in the number of affected vessels and in the incidence of moderate stenosis (3,8). Detection of narrowing of the coronary lumen in patients with diabetes is often impaired by autonomic neuropathy (9,10), which can reduce the symptoms of ischemic CHD, delay its detection, and worsen the prognosis. In addition, diabetic individuals are faced with increased restenosis and mortality rates following revascularization procedures, especially for percutaneous transluminal coronary angioplasty (PTCA) (11). The impact of CHD on myocardial function in diabetes is exacerbated by diabetic cardiomyopathies (12,13), which can impair cardiac contractile function and may accelerate heart failure. Multivariate analyses of a number of large prospective studies with follow-up of 12 to 20 years, including the Framingham Study, the Multiple Risk Factor Intervention Trial,
and the Nurses’ Health Study, have demonstrated that diabetes is associated with two- to fivefold increases in CHD- and cardiovascular-related death (14,15,16). This CVD accounts for the majority of premature mortality associated with both type 1 and type 2 diabetes worldwide (17). A combination of factors related to hyperglycemia, hypertension, insulin resistance, dyslipidemia, hypercoagulability, and inflammation contributes to the etiology of CVD associated with diabetes.
and the Nurses’ Health Study, have demonstrated that diabetes is associated with two- to fivefold increases in CHD- and cardiovascular-related death (14,15,16). This CVD accounts for the majority of premature mortality associated with both type 1 and type 2 diabetes worldwide (17). A combination of factors related to hyperglycemia, hypertension, insulin resistance, dyslipidemia, hypercoagulability, and inflammation contributes to the etiology of CVD associated with diabetes.
HISTOLOGIC CLASSIFICATION OF ATHEROSCLEROSIS
Atherosclerosis is a progressive disease of the arterial wall involving components of inflammation, vascular lipid deposition and remodeling, fibrosis, and thrombosis. The American Heart Association has outlined a histologic classification of atherosclerotic lesions (Table 52.1) (18,19). Initial atherosclerotic lesions and fatty streaks represent the appearance of scattered lipid deposits in the intima associated with increases in macrophages and macrophage foam cells. Although these early lesions are not clinically significant and may regress (20), they also can provide precursors for the formation of advanced complex plaques. Intermediate lesions and atheroma involve increased focal accumulation of lipid within the neointima due to increased levels of foam cells, of both macrophage and vascular smooth muscle cell (VSMC) origin, and the appearance of extracellular lipid droplets, leading to the formation of a lipid-rich plaque core. These intermediate lesions appear to arise from either fatty streaks or preexisting intimal cell masses (21). Fibrous plaques are formed as a cap of connective tissue, mainly involving VSMC in a collagen and proteoglycan matrix, and accumulate between the lipid core and vessel lumen. Thickness of the fibrous cap and its infiltration with macrophages affect the stability of these lesions, such that plaques with thin fibrous caps and those with increased levels of macrophages appear prone to rupture (3,22). Fibrotic lesions also can develop in plaques that do not display a well-defined lipid core. Complicated lesions occur when the plaque becomes disrupted owing to fissure, hemorrhage, or thrombus formation. Recent proposed modifications of this classification have emphasized that fibrous plaques or pathologic intimal thickenings can lead to significant stenosis without thrombi and that luminal thrombosis often occurs in vessels with a less than 50% reduction in diameter (23). Moreover, atherogenesis does not necessarily progress in a uniform manner, but lesion histology instead involves a mosaic of accumulations of lipid, fibrous material, and cellular mass.
TABLE 52.1. Histologic Classification of Atherosclerosis | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Recent analyses of coronary tissue from autopsy studies have indicated that the main causes of coronary sudden death are plaque rupture or erosion, followed by thrombosis and stenosis (23). Plaque rupture is evidenced by contact between the thrombus and the necrotic atheromatous core. Plaque erosion is indicated when the thrombus occurs on a fibrous cap that has lost its luminal endothelium but retains a continuous connective tissue covering the necrotic core. Disruption of the endothelium and fibrous cap can expose the blood to the plaque
core rich in tissue factor (24,25), which can trigger the extrinsic coagulation pathway. Moreover, fibrin-rich thrombi may be stabilized by plasminogen activator inhibitor-1 (PAI-1), either circulating or locally released from neointimal smooth muscle cells (SMC) (26,27). Although thrombosis is observed in the majority of cases of coronary sudden death, approximately 20% of these coronary events occur by luminal narrowing without intramural thrombosis (23). Multiple mechanisms lead to narrowing of the coronary lumen, including the progressive expansion of the lesion mass due to lipid accumulation, fibrosis, and neointimal SMC growth; intraplaque hemorrhage; healed plaque erosions and thrombi; and contracture caused by SMC constriction and vascular wound healing (28,29).
core rich in tissue factor (24,25), which can trigger the extrinsic coagulation pathway. Moreover, fibrin-rich thrombi may be stabilized by plasminogen activator inhibitor-1 (PAI-1), either circulating or locally released from neointimal smooth muscle cells (SMC) (26,27). Although thrombosis is observed in the majority of cases of coronary sudden death, approximately 20% of these coronary events occur by luminal narrowing without intramural thrombosis (23). Multiple mechanisms lead to narrowing of the coronary lumen, including the progressive expansion of the lesion mass due to lipid accumulation, fibrosis, and neointimal SMC growth; intraplaque hemorrhage; healed plaque erosions and thrombi; and contracture caused by SMC constriction and vascular wound healing (28,29).
CHARACTERISTICS OF CORONARY ATHEROSCLEROTIC LESIONS IN DIABETES
Histopathologic comparison of coronary atherectomy specimens from nondiabetic and diabetic subjects has revealed an increase in the percentage of the total area occupied by lipid-rich atheromatous tissue from 2% ± 1% to 7% ± 2% (p = 0.01) and an increase in the incidence of coronary thrombus from 40% to 62% (p = 0.04) for nondiabetics and diabetics, respectively (3). Coronary artery atherectomy specimens from subjects with type 2 diabetes have a twofold increase in PAI-1 antigen and a decrease in the level of immunoreactive urokinase plasminogen activator, compared with specimens from age- and gender-matched controls without diabetes (30). Macrophage infiltration was also elevated by nearly twofold (p = 0.003) in atherectomy specimens from diabetic subjects (3). In contrast, morphometric analysis of coronary atherectomy specimens from primary lesions has indicated that the relative amounts of collagen-rich and hypercellular plaque components, which comprised approximately 90% of the total lesion area, were similar in diabetic and nondiabetic subjects (3,30). However, given the nature of tissue isolation by directional coronary atherectomy, the cellularity of these specimens in these studies may not represent possible effects of diabetes on SMC hyperplasia at the base or peripheral areas of the stenotic lesions.
Coronary artery calcification in diabetic subjects appears either similar (31) or increased (32,33), compared with that in nondiabetic control subjects. Although plaque calcification can provide a marker of plaque burden and is predictive of future coronary events (34,35), the effects of calcification on the pathophysiology of atherosclerotic lesions are not fully understood. Calcium deposition does not appear to adversely affect plaque mechanical stability (36); however, arterial calcification can increase stiffening, resulting in reduced vascular compliance.
EFFECTS OF DIABETES ON THE PATHOGENESIS OF ATHEROSCLEROSIS
Combinations of pathophysiologic processes contribute to the development of atherosclerosis and its sequel of stenosis and coronary sudden death. As described above, atherosclerotic lesions can arise from fatty streaks or neointimal thickenings. These lesions can expand progressively to reduce luminal diameter and can undergo rupture or erosion, which can trigger thrombosis (23). The atherogenic effects of diabetes and insulin resistance appear to be initiated by a combination of metabolic abnormalities related to hyperglycemia, impaired insulin action or insulin deficiency, a proatherogenic dyslipidemia, and confounding factors such as hypertension and obesity (Fig. 52.1). In addition, vasoactive hormones, cytokines, and growth factors, including angiotensin II (ATII), tumor necrosis factor-α (TNF-α), and vascular endothelial growth factor (VEGF) amplify and in part mediate the adverse vascular effects of these metabolic abnormalities. These metabolic and hormonal imbalances can induce endothelium dysfunction, vascular inflammation, SMC growth, intimal lipid accumulation, fibrosis, and hypercoagulability, leading to atherosclerosis and thrombosis (Fig. 52.1). The effects of diabetes and insulin resistance on a number of these proatherogenic pathways are described further on.
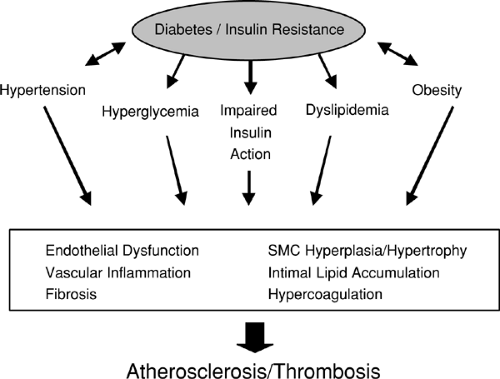 Figure 52.1. Diabetes, insulin resistance, and cardiovascular disease. Diabetes and insulin resistance are associated with a combination of metabolic abnormalities such as hyperglycemia, insulin resistance, insulin deficiency, hyperinsulinemia, hypertriglyceridemia, and reduced high-density lipoprotein that contribute to vascular dysfunctions, atherosclerosis, and thrombosis. In addition, diabetes and insulin resistance often coexist with additional cardiovascular risk factors, including hypertension and obesity, as the insulin resistance syndrome. SMC, smooth muscle cells. |
Endothelial-Leukocyte Adhesion
Initial lesions and fatty streaks, also described as intimal xanthoma (23), are characterized by the appearance of macrophage foam cells in the subendothelial area. These foam cells arise from circulating monocytes that have adhered and infiltrated into the vessel wall, differentiated into macrophages, and become lipid laden via the avid uptake of modified low-density lipoprotein (LDL). Diabetes and insulin resistance promote leukocyte infiltration into the arterial wall via several mechanisms. Diabetes and hyperglycemia increase expression of E-selectin, vascular cell adhesion molecule-1 (VCAM-1), and intercellular adhesion molecule-1 (ICAM-1) by endothelial cells (37,38,39), which bind to leukocyte glycoprotein counterreceptors and integrins, thereby mediating leukocyte tethering, rolling, and firm adhesion (Fig. 52.2). In addition, leukocytes isolated from diabetic subjects display increased adhesiveness to endothelial cells (40,41). Following endothelial-leukocyte adhesion, infiltration of leukocytes to the subendothelial interstitium is stimulated by monocyte chemoattractant protein-1 (MCP-1), which is upregulated in diabetes (42). The infiltrated monocytes differentiate into macrophages, which produce a number of proatherogenic effects by accumulating lipids (43,44) and releasing proinflammatory cytokines and matrix metalloproteinases (45,46,47). The elaboration of cytokines, growth factors, metalloproteases, and procoagulants from these macrophages likely promotes plaque expansion and generates plaques prone to rupture and thrombosis.
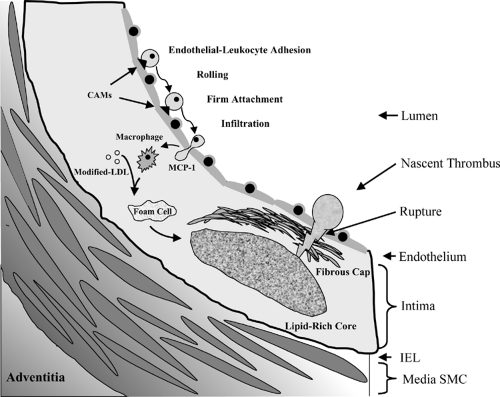 Figure 52.2. Components contributing to the development of fibrous-cap atheroma. Leukocyte binding to endothelial cell adhesion molecules (CAMs) and chemoattraction by monocyte chemoattractant protein-1 (MCP-1) increase the infiltration of monocytes into the subendothelial intima. These monocytes differentiate into macrophages, which release proinflammatory cytokines and endocytose modified low-density lipoprotein to generate foam cells. Focal accumulation of intimal lipid in the form of increased foam cells and extracellular lipid deposits results in the formation of a lipid-rich plaque core, which can become necrotic. Plaques also accumulate fibrotic material consisting of a collagen-proteoglycan matrix and smooth muscle cells (SMC) that have migrated across the internal elastic lamina (IEL). Rupture of this fibrous cap over the lipid core exposes this atheromatous material rich in tissue factor to circulating factors VII and VIIa, which initiates thrombogenic cascades and the formation of a thrombus. |
Formation of Lipid-Rich Lesions
Monocyte-derived macrophages endocytose modified LDL via multiple receptors, including both class A and class B scavenger
receptors (43,44), lectin-like oxidized LDL receptor-1 (48), and lipoprotein lipase (49). Uptake of lipid in excess of its metabolism results in the formation of macrophage foam cells laden in cholesterol esters. In addition, foam cells can develop similarly from VSMC by the uptake of oxidized LDL via class A and class B scavenger receptors (50,51). Diabetes exacerbates the uptake of modified LDL by foam cells via several mechanisms. First, hyperglycemia increases glucoxidation, resulting in increased levels of oxidized and glycated LDL (52,53), which increase ligand availability for scavenger receptors and lipoprotein lipase. Second, diabetes increases the expression of macrophage class B scavenger receptor CD36 (54), which enhances oxidized LDL endocytosis. Third, diabetes causes a proatherogenic dyslipidemia resulting in reduced high-density lipoprotein (HDL) levels and increased triglycerides, which can reduce reverse cholesterol transport and contribute to increased levels of small dense LDL particles (55). In addition, macrophage foam cells release proinflammatory cytokines, such as TNF-α and interleukin-1, which can induce apoptosis of endothelial cells and VSMC (56). Focal accumulation of foam cells, formation of extracellular lipid droplets, apoptosis of VSMC, and disruption of vascular smooth muscle architecture can result in the formation of a necrotic lipid-rich core (Fig. 52.2).
receptors (43,44), lectin-like oxidized LDL receptor-1 (48), and lipoprotein lipase (49). Uptake of lipid in excess of its metabolism results in the formation of macrophage foam cells laden in cholesterol esters. In addition, foam cells can develop similarly from VSMC by the uptake of oxidized LDL via class A and class B scavenger receptors (50,51). Diabetes exacerbates the uptake of modified LDL by foam cells via several mechanisms. First, hyperglycemia increases glucoxidation, resulting in increased levels of oxidized and glycated LDL (52,53), which increase ligand availability for scavenger receptors and lipoprotein lipase. Second, diabetes increases the expression of macrophage class B scavenger receptor CD36 (54), which enhances oxidized LDL endocytosis. Third, diabetes causes a proatherogenic dyslipidemia resulting in reduced high-density lipoprotein (HDL) levels and increased triglycerides, which can reduce reverse cholesterol transport and contribute to increased levels of small dense LDL particles (55). In addition, macrophage foam cells release proinflammatory cytokines, such as TNF-α and interleukin-1, which can induce apoptosis of endothelial cells and VSMC (56). Focal accumulation of foam cells, formation of extracellular lipid droplets, apoptosis of VSMC, and disruption of vascular smooth muscle architecture can result in the formation of a necrotic lipid-rich core (Fig. 52.2).
Fibrous-Cap Formation
Atherosclerotic lesions accumulate fibrous material consisting mainly of VSMC in a collagen-proteoglycan matrix. The fibrous material that separates the plaque core from the luminal surface of the vessel is termed the fibrous cap (Fig. 52.2). Because the atheromatous material is thrombogenic in part attributable to high levels of tissue factor (24,25), the fibrous cap provides a barrier to thrombosis by separating this pool of tissue factor from the blood. The specific effects of diabetes on the composition and stability of the fibrous cap in primary lesions have not yet been described. Diabetes can increase SMC proliferation and extracellular matrix (ECM) production in a porcine atherosclerosis model (57) and increases neointimal hyperplasia and the accumulation of collagen-rich sclerotic tissue in restenotic lesions (58,59). Although these findings suggest that diabetes can promote the accumulation of fibrous material in restenotic lesions, it is unclear whether these findings can be extrapolated to characteristics of the fibrous cap. Several of the effects of diabetes on plaque composition may influence fibrous-cap formation and stability. Increased matrix metalloproteinase activity and macrophage levels in diabetic lesions (60) may shift the proteolytic balance within the cap, leading to the degradation of ECM and weakening of its structural integrity (61), thus increasing susceptibility to rupture or erosion. As described previously, PAI-1 levels are also increased in atherectomy specimens in diabetes. Elevated levels of PAI-1 can reduce SMC migration (62) and thereby might decrease incorporation of SMC into the fibrous cap. Alternatively, increased PAI-1 within lesions also may reduce plasmin-stimulated ECM turnover and thereby promote fibrosis (63). These effects of PAI-1 may lead to the generation of fibrous material with reduced cellularity and increased ECM.
Atherothrombosis
Thrombosis is the major cause of coronary stenosis and sudden death (23). Intramural arterial thrombosis can be triggered by the disruption of complex plaques, which expose the blood to thrombogenic factors that are enriched in the subendothelial area. Tissue factor, the major activator of the extrinsic coagulation pathway, is highly expressed in the plaque core (24,25). Disruption of the plaque surface allows this tissue factor to bind circulating factor VII in blood, resulting in the generation of active VIIa and the formation of tissue factor-VIIa complexes. This complex leads to a cascade involving activation of factor X, formation of prothrombinase complex, generation of thrombin, and cleavage of fibrinogen to fibrin. Elevated levels of tissue factor in vascular tissues in diabetic and obese/insulin-resistant murine models (64,65) may predispose diabetic arteries to the activation of this coagulation cascade. Moreover, increased arterial expression of PAI-1 in diabetes may impair the dissolution of nascent thrombi (30,66). Fibrin-rich thrombi that develop on ruptured plaques can expand to cause occlusion, can spontaneously undergo dissolution, or may become lined with endothelium and incorporated as an expansion of the neointima (28). Although atherosclerotic lesions in diabetes can have attributes (increased PAI-1 and macrophages) that would be expected to promote thrombosis, the direct effects of diabetes on atherothrombosis have not yet been established. A recent report, however, has provided evidence for an increase in healed ruptures in coronary sudden death in diabetes (glucose intolerance) (28), suggesting that diabetes is associated with an increased occurrence of ruptures that contribute to subclinical plaque burden.
Restenosis following Revascularization
The Bypass Angioplasty Revascularization Investigation (BARI) demonstrated that patients with treated diabetes who underwent PTCA or coronary artery bypass graft surgery (CABG) had cardiac-related mortality rates of 20.6% or 5.8%, respectively, after an average follow-up of 5.4 years (67). In contrast, the cardiac mortality rates for other BARI patients receiving PTCA and CABG were 4.8% and 4.7%, respectively. At 7 years of follow-up of BARI patients, the survival of the group with treated diabetes was 76.4% for CABG and 55.7% for PTCA (p = 0.0011) compared with survival rates of 86.4% (CABG) and 86.8% (PTCA) for patients without diabetes (11). The basis for the 3.5-fold higher mortality rates for diabetic patients receiving PTCA compared with their nondiabetic counterparts is not fully understood. Serial intravascular ultrasound has indicated that increased restenosis following coronary interventions in diabetes is due to intimal expansion (58). Analysis of coronary atherectomy specimens from restenotic lesions after PTCA revealed that the fraction of collagen-rich sclerotic tissue was increased in lesions from patients with diabetes (59). These results suggest that diabetes increases neointimal fibrosis of arterial lesions resultant from PTCA. Diabetes has also been shown to increase the incidence of restenosis following arterial stenting and saphenous vein graft bypass (68,69,70).
Animal models have been utilized to examine the effect of diabetes on the neointimal response to balloon catheterization. Balloon catheter-induced injury causes endothelial denudation and arterial stretch, resulting in the formation of a neointima from the proliferative expansion of VSMC, which have migrated across the internal elastic lamina from the vessel media. Streptozotocin (STZ)-induced diabetes in rats has been shown to reduce carotid artery neointimal formation by approximately 50% compared with that in nondiabetic and insulin-treated diabetic controls in this balloon-injury model (71,72). Similarly, alloxan-induced diabetes reduced balloon catheter-induced neointimal formation in rabbits (73). STZ-induced diabetes did not affect the neointimal thickening in response to coronary stent placement in Yucatan miniature swine (74). However, diabetes was associated with increased in-stent thrombosis resulting in sudden death in this porcine model (74). These findings
indicate that hyperglycemia caused by STZ-induced diabetes does not increase neointimal hyperplasia but may increase thrombosis in injured vessels. The effects of insulin resistance and type 2 diabetes on the balloon injury-induced neointimal response have been examined in rat obesity models. These studies reveal that neointimal hyperplasia was increased in the type 2 diabetic/insulin-resistant obese Zucker rats and Otsuka Long-Evans Tokushima fatty rats compared with their lean insulin-sensitive controls (72,75,76).
indicate that hyperglycemia caused by STZ-induced diabetes does not increase neointimal hyperplasia but may increase thrombosis in injured vessels. The effects of insulin resistance and type 2 diabetes on the balloon injury-induced neointimal response have been examined in rat obesity models. These studies reveal that neointimal hyperplasia was increased in the type 2 diabetic/insulin-resistant obese Zucker rats and Otsuka Long-Evans Tokushima fatty rats compared with their lean insulin-sensitive controls (72,75,76).
The interpretation of these animal studies is beset with a number of limitations. First, balloon injuries were performed on vessels with little or no preexisting vascular disease. This contrasts with clinical situations, in which revascularization is performed on restenotic vessels with advanced plaque pathologies. Second, most studies that have examined the effect of diabetes on atherosclerosis and neointimal hyperplasia in animal models have utilized the islet β-cell cytotoxins, including STZ or alloxan, to reduce insulin production. Although this approach is effective in generating hyperglycemia resulting from insulin deficiency, these models do not replicate the etiology of type 1 diabetes mediated via autoimmune destruction of β-cells. Third, rat models with obesity-induced insulin resistance have multiple endocrine abnormalities, which may contribute to the exaggerated neointimal response. Although the effects of diabetes on vascular proliferation are not fully understood, neointimal smooth muscle proliferation represents a major component of atherogenesis and an important target for emerging therapeutic strategies (77).
ENDOTHELIAL DYSFUNCTIONS IN DIABETES
The endothelium plays an integral role in vascular structure, hemodynamics, and hemostasis. Diabetes has a number of adverse effects on the vascular endothelium that impair these functions and likely facilitate atherosclerosis and thrombosis. As described above, diabetes increases the expression of leukocyte adhesion molecules that mediate binding and the infiltration of monocytes into the arterial subendothelial space. In addition, diabetes impairs endothelial barrier function, which increases transendothelial permeability and access of circulating molecules to the vascular interstitium. Diabetes also alters endothelial cell synthesis of a host of vasoactive hormones and factors that affect vascular tone, hemodynamics, coagulation, and VSMC physiology. The effects of diabetes on several key endothelial functions are described below.
Permeability of Endothelial Tight Junctions
Increased endothelial permeability is a hallmark of vascular dysfunction in diabetes (78,79). This increased permeability may facilitate penetration of proatherogenic cytokines and lipoproteins into the subendothelial space. Several mechanisms have been postulated to explain the increased transport of molecules across the endothelium in diabetes, including enhanced pinocytosis (78) and increased intercellular diffusion caused by reduced tight-junction formation (80). Diabetes can impair endothelium barrier function via the increased production of VEGF (also called vascular permeability factor) by VSMC. In this pathway, hyperglycemia and hormones, such as ATII, increase synthesis and secretion of VEGF in arterial SMC (81,82). This VEGF can act in a paracrine manner on endothelial cells, where it binds kinase insert domain-containing receptors VEGF-R2 (KDR) and stimulates phosphorylation of tight-junction proteins, such as zonula occluden 1 (83). However, although increased VEGF levels in diabetes have been shown in certain tissues, such as the retina (84), less is known regarding the effects of diabetes on VEGF expression in cardiovascular tissues. In addition, hyperglycemia can exert additional effects on the endothelium via the activation of protein kinase C (PKC) and the increased formation of advanced glycation end-products (AGEs) and reactive oxygen species, which can act directly on endothelial cells to induce hyperpermeability (79,85).
Endothelium-Dependent Vasorelaxation
The vascular endothelium releases several factors that induce vascular smooth muscle relaxation, including nitric oxide (86), prostacyclin I2, and one or more endothelium-derived hyperpolarizing factors (87). Of these factors, impaired endothelium-dependent vasorelaxation in diabetes has been attributed primarily to the reduced bioavailability of nitric oxide. Nitric oxide production by endothelial nitric oxide synthase (eNOS) is increased in response to shear stress and potent vasodilator hormones such as acetylcholine and bradykinin (86,88) (Fig. 52.3). Nitric oxide emanating from the endothelium acts in a paracrine manner on the vascular smooth muscle, where it binds to soluble guanylyl cyclase and thereby increases the synthesis of cyclic guanosine monophosphate (cGMP) (Fig. 52.3). Elevated cGMP levels activate cGMP-dependent protein kinases, including cGMP kinase I, which play an important role in regulating cytosolic calcium homeostasis. cGMP kinase I phosphorylates the microsomal inositol 1,4,5-trisphosphate (IP3) receptor complex (89) and thereby inhibits IP3-stimulated calcium release from intracellular stores, resulting in SMC relaxation.
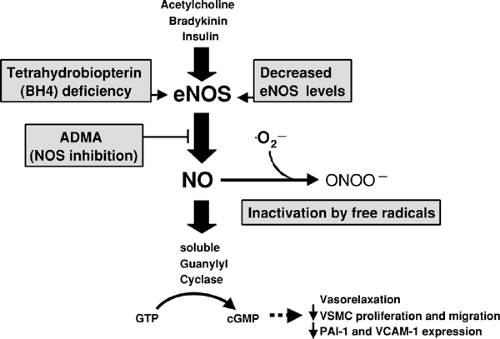 Figure 52.3. Mechanisms responsible for reduced nitric oxide (NO) bioactivity in diabetes. Mechanisms that contribute to reduced NO production include reduced endothelial nitric oxide synthase (eNOS) expression, tetrahydrobiopterin deficiency, and competitive inhibition of eNOS catalytic activity by elevated endogenous asymmetric dimethylarginine (ADMA). In addition, bioactivity of NO can also be reduced by the oxidation of NO to peroxynitrite (ONOO–). VSMC, vascular smooth muscle cells; PAI-1, plasminogen activator inhibitor-1; VCAM-1, vascular cell adhesion molecule-1; GTP, guanosine triphosphate; cGMP, cyclic guanosine monophosphate. |
Reduced vasodilation response to infused acetylcholine (or methacholine, a long-acting muscarinic receptor agonist) and reactive hyperemia have been demonstrated in both type 1 and type 2 diabetes (90,91,92,93). Acute hyperglycemia, induced by hyperglycemic clamp, maintained at 300 mg/dL glucose levels for 6 hours, impairs methacholine-induced vasorelaxation in healthy nondiabetic subjects (94). However, although an acute increase in glucose can impair endothelial function, it is unclear whether the mechanism responsible for this effect of acute hyperglycemia is related to the chronic effects of hyperglycemia associated with diabetes. Impaired endothelium-dependent relaxation also occurs in insulin-resistant patients in the absence of overt type 2 diabetes (90,95). Thus, endothelial dysfunction in type 2 diabetes may reflect the combined adverse effects of hyperglycemia and additional metabolic and hormonal abnormalities associated with insulin resistance, such as an increase in free fatty acids and reduced insulin action (96,97). Results from studies of a variety of experimental animal models of type 1 diabetes have demonstrated that reduced endothelium-dependent relaxation occurs in ex vivo preparations of coronary, mesenteric, and carotid arteries and aorta (98,99,100,101), indicating that this dysfunction is retained in isolated vessels. Because most reports have shown a normal vasodilator response to nitric oxide donors such as sodium nitroprusside (91,102,103), the impairment of vasodilation in diabetes appears to be due mainly to an impairment in endothelium-derived nitric oxide. However, some reports have shown that vasodilator response to nitric oxide donors is also impaired in diabetes and insulin resistance (90,104), suggesting that in certain situations impaired endothelium-dependent vasorelaxation may be superimposed on impaired endothelium-independent relaxation. The mechanism or mechanisms responsible for this resistance to vasorelaxation by nitric oxide donors have not yet been defined. Potential mechanisms could be related to chronic vasoconstriction or oxidant production caused by endothelin-1 or ATII (105,106,107).
Reductions in both nitric oxide synthesis and stability have been proposed to explain impaired endothelium-dependent
nitric oxide-mediated vasodilation in diabetes. Increased reactive oxygen species in diabetes have been suggested to decrease the half-life of nitric oxide by converting it to peroxynitrite (Fig. 52.3). Consistent with this theory, elevated vascular superoxide production in diabetes via NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) oxidase has been demonstrated 101,108), and antioxidant treatments, including vitamin C, have been shown to improve endothelium-dependent vasorelaxation in diabetes (92,109). Moreover, impairment of endothelial NADPH oxidase activity by the targeted mutation of the gp91phox gene has been shown to reduce arterial oxygen-radical formation and enhance endothelium-dependent vasorelaxation (110). Although there is considerable molecular and pharmacologic evidence for a role of vascular oxidants in attenuating endothelium-derived nitric oxide action, potential clinical benefits of antioxidant supplementation, such as vitamin E (RRR-α-tocopheryl acetate at 400 IU/day), for cardiovascular outcomes have not yet been identified (111). However, the potential benefits of substantially higher doses of vitamin E (1,200 IU/day), which may reduce monocyte activity and levels of soluble cell adhesion molecules (112), for cardiovascular complications remain to be examined.
nitric oxide-mediated vasodilation in diabetes. Increased reactive oxygen species in diabetes have been suggested to decrease the half-life of nitric oxide by converting it to peroxynitrite (Fig. 52.3). Consistent with this theory, elevated vascular superoxide production in diabetes via NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) oxidase has been demonstrated 101,108), and antioxidant treatments, including vitamin C, have been shown to improve endothelium-dependent vasorelaxation in diabetes (92,109). Moreover, impairment of endothelial NADPH oxidase activity by the targeted mutation of the gp91phox gene has been shown to reduce arterial oxygen-radical formation and enhance endothelium-dependent vasorelaxation (110). Although there is considerable molecular and pharmacologic evidence for a role of vascular oxidants in attenuating endothelium-derived nitric oxide action, potential clinical benefits of antioxidant supplementation, such as vitamin E (RRR-α-tocopheryl acetate at 400 IU/day), for cardiovascular outcomes have not yet been identified (111). However, the potential benefits of substantially higher doses of vitamin E (1,200 IU/day), which may reduce monocyte activity and levels of soluble cell adhesion molecules (112), for cardiovascular complications remain to be examined.
Diabetes has also been reported to reduce vascular nitric oxide synthesis (98). Multiple mechanisms have been proposed to explain the decreased eNOS activity in diabetes and insulin resistance. Reduced eNOS expression has been described in adipose microvessels isolated from obese insulin-resistant Zucker rats and coronary microvessels from alloxan-induced diabetic dogs (98,113), suggesting that reduced protein levels of eNOS may contribute to lower nitric oxide production. In addition, the elevation of circulating levels of asymmetric dimethylarginine, an endogenous NOS inhibitor (114), and a deficiency in tetrahydrobiopterin (115,116), a cofactor for eNOS, have also been implicated in contributing to reduced nitric oxide generation in diabetes and insulin resistance.
The implications of reduced endothelium-derived nitric oxide bioactivity in diabetes goes beyond its hemodynamic effects. Nitric oxide, via cGMP-dependent pathways, has been shown to suppress a number of atherogenic and thrombogenic processes within the vascular wall, including neointimal SMC migration and proliferation and endothelial-leukocyte adhesion (117) (Fig. 52.2). Consistent with these possible antiatherogenic actions of nitric oxide, eNOS deficiency has been reported to increase atherosclerotic lesion size in apolipoprotein E (ApoE)-deficient mice (118). In addition, nitric oxide can also suppress the expression of thrombogenic factors, such as PAI-1 and tissue factor (119,120), which may shift the coagulation balance away from atherothrombosis.
EFFECTS OF DIABETES ON VASCULAR COMPLIANCE AND ARTERIAL STIFFNESS
Arterial stiffness is strongly associated with atherosclerosis (121). The Strong Heart Study (12) and the Atherosclerosis Risk in Communities Study (122) have reported increased arterial stiffness and reduced compliance in subjects with type 2 diabetes. Arterial compliance is also reduced in individuals with type 1 diabetes (123,124). Several theories have been proposed to explain the increased arterial stiffening in diabetes. Increased glycation of ECM proteins, such as collagen and elastin, may increase covalent intermolecular cross-linking and thereby reduce elasticity. Consistent with this theory, treatment with an AGE cross-link “breaker” has been shown to reverse diabetes-induced arterial stiffness (125). Increased arterial stiffness may also result from arterial calcification, arterial-wall thickening, or increased chronic VSMC contraction due to an imbalance in vasoactive hormone activities. Impaired arterial elasticity might contribute to atherogenesis by increasing mechanical strain and shear forces, causing endothelial damage, activation of stretch-activated mechanoreceptors, and increased release of trophic factors (126,127,128).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


