Diabetic Ketoacidosis and Hyperosmolar Hyperglycemic State
Jennifer Wyckoff
Martin J. Abrahamson
DEFINITIONS
Hyperosmolar hyperglycemic state (HHS) and diabetic ketoacidosis (DKA) represent two distinct metabolic derangements manifested by insulin deficiency and severe hyperglycemia. HHS occurs when insulin deficiency relative to insulin requirements causes hyperglycemia, which in turn leads to dehydration, ultimately resulting in a severe hyperosmolar state. DKA occurs in the setting of more severe insulin deficiency, when low circulating levels of insulin lead not only to hyperglycemia and dehydration but also to the production of ketone bodies and acidosis. DKA is defined as the presence of all three of the following: (i) hyperglycemia (glucose >250 mg/dL), (ii) ketosis, and (iii) acidemia (pH <7.3) (1). HHS is characterized by severe hyperglycemia and hyperosmolarity. HHS and DKA are not mutually exclusive but rather two conditions that both result from some degree of insulin deficiency. They can and often do occur simultaneously. In fact, one third of patients admitted for hyperglycemia exhibit characteristics of both HHS and DKA (2).
EPIDEMIOLOGY
In one of the largest studies of the epidemiology of DKA, Faich et al. (3) reported an annual incidence of DKA in Rhode Island of 46 cases per 10,000 persons with diabetes and of 1.4 cases per 10,000 persons in the general population. Nationally, DKA contributes to approximately 100,000 hospital admissions per year (1,4,5,6) and accounts for 2% to 9% of hospital admissions in persons with diabetes (1,3,7,8).
More than 20% of patients admitted for DKA had previously undiagnosed diabetes (3,9,10). Another 15% of admissions were of patients with multiple admissions for DKA (3). Several studies reported that the average age of patients admitted for DKA was 40 to 50 years (3,4), but that the risk decreased with age (10). Some studies have reported a female predominance (3,6,9,10), possibly because young women were more likely to have repeated episodes of DKA (9,11). A slight increase in cases during the winter months has also been cited (3,9).
DKA is the leading cause of death of patients with diabetes who are younger than 24 years old, accounting for about one half of the deaths in this population group (5). Before the discovery of insulin in 1922, the mortality due to DKA was virtually 100% (12). By 1932, mortality decreased to 29% (12). By the 1950s, the reported mortality was 15%, with the improvement credited primarily to the widespread use of antibiotics, the introduction of intravenous potassium replacement, and the use of norepinephrine for blood pressure support (13). Mortality rates ranging from 2.5% to 9% among patients admitted with DKA have been reported in more recent studies (3,5,9,12). The
studies reporting all-cause in-hospital mortality reported higher mortality rates than studies that reported mortality due to DKA alone and excluded deaths attributable to the factors that precipitated the DKA (11). Mortality among patients with DKA has been related to age, level of consciousness on admission, severity of acidosis, degree of hyperosmolarity, severity of azotemia, and nursing home residence (2,3,10). In addition to short-term mortality, there appears to be an increased risk of microvascular complications of diabetes with long-term follow-up of survivors of DKA (10).
studies reporting all-cause in-hospital mortality reported higher mortality rates than studies that reported mortality due to DKA alone and excluded deaths attributable to the factors that precipitated the DKA (11). Mortality among patients with DKA has been related to age, level of consciousness on admission, severity of acidosis, degree of hyperosmolarity, severity of azotemia, and nursing home residence (2,3,10). In addition to short-term mortality, there appears to be an increased risk of microvascular complications of diabetes with long-term follow-up of survivors of DKA (10).
One of the most interesting aspects of the epidemiologic study of hyperglycemic states is the epidemiology of DKA in various ethnic groups. In a retrospective chart review of adults with DKA admitted between 1994 and 1995 to a Texas hospital, 80% of whites admitted with DKA were classified as having type 1 diabetes (14). In contrast, only 53% of African Americans and 34% of Hispanic patients who were admitted with DKA had type 1 diabetes. By definition, patients who required insulin from the time of diagnosis of diabetes were considered to have type 1 diabetes, and patients who had prolonged treatment with either diet or oral hypoglycemic agents were considered to have type 2 diabetes. No difference was found in serum electrolytes, renal function, glucose, pH, anion gap, osmolality, or level of ketosis between those classified as having type 1 diabetes and those classified as having type 2 diabetes. Despite the conventional wisdom that DKA in type 2 diabetes usually is triggered by extreme physiologic stress, in the majority of these cases, no precipitating event could be identified.
PATHOPHYSIOLOGY
Understanding the clinical manifestations of DKA and HHS requires a thorough knowledge of the pathophysiology of these two closely related disturbances. In both DKA and HHS, relative insulin deficiency is the critical underlying defect. In both DKA and HHS, inadequate levels of circulating insulin relative to insulin requirements lead to hyperglycemia, which in turn leads to dehydration. Hyperglycemia can lead to progressive dehydration and hyperosmolarity and ultimately to HHS. If insulin deficiency is severe enough, ketosis and ultimately acidosis develop (Fig. 53.1).
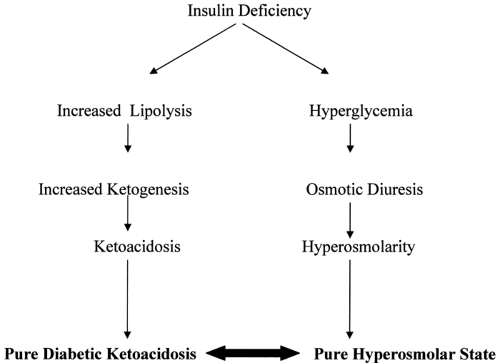 Figure 53.1. Hyperglycemic states. |
Relative insulin deficiency—not absolute insulin deficiency—is necessary for the development of both DKA and HHS. Even patients with type 2 diabetes and “normal” insulin levels may develop DKA if the level of insulin resistance causes a large enough increase in insulin requirements. Insulin resistance is mediated by several factors, including the underlying pathophysiology of type 2 diabetes and an increase in the levels of counterregulatory hormones, including cortisol, glucagon, epinephrine, and growth hormone (Table 53.1).
TABLE 53.1. Regulation of Ketogenesis and Glucose Metabolism | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
There is an extensive literature exploring the relative importance of insulin deficiency and insulin resistance due to counterregulatory hormones to the development of DKA (15,16,17,18,19,20,21,22,23,24). The facts that the incidence of DKA has decreased dramatically since the introduction of insulin in the 1920s and that insulin administration is the mainstay of therapy for DKA demonstrate the central role of insulin deficiency in the development of DKA (15). Early studies supporting insulin deficiency as the primary cause of DKA demonstrated low levels of insulin or C-peptide or both in the face of hyperglycemia (15,16). Withdrawal of insulin from patients with type 1 diabetes results in hyperglycemia and increased levels of ketone bodies within a matter of hours, as well as in elevations in some counterregulatory hormones (17).
Despite this evidence, the impact of increased insulin resistance due to the counterregulatory hormones in the promotion of DKA and HHS must not be underestimated. Clinicians appreciate the increased risk of DKA that occurs with any physiologic stress in the patient with type 1 diabetes. Elevated levels of counterregulatory hormones have been observed during episodes of DKA (18,19,20,21). Furthermore, the severity of DKA appears to be decreased in conditions in which there is a deficiency of counterregulatory hormones (22,23).
Nevertheless, in the absence of insulin deficiency, elevated levels of counterregulatory hormones alone do not cause DKA. Gerich et al. (24) studied the effects of infusing counterregulatory hormones into insulin-dependent patients. Neither glucagon
infusion nor growth hormone infusion produced an elevation in free fatty acids or ketone bodies until insulin was withdrawn.
infusion nor growth hormone infusion produced an elevation in free fatty acids or ketone bodies until insulin was withdrawn.
HYPERGLYCEMIA
Hyperglycemia occurs in both HHS and DKA. Hyperglycemia results from increased glucose production (gluconeogenesis and glycogenolysis) and decreased peripheral utilization (glycolysis, lipogenesis, and glycogen synthesis). Figure 53.2 gives an overview of these processes. Cox and Nelson (25) provide an excellent comprehensive review of the biochemistry involved in hyperglycemia.
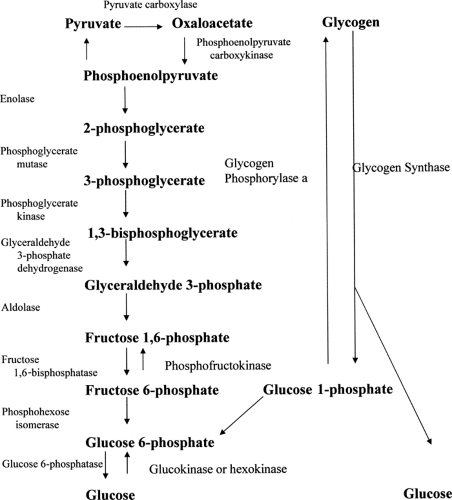 Figure 53.2. Ketogenesis. |
Gluconeogenesis refers to the production of glucose from pyruvate or oxaloacetate. Glycolysis refers to the inverse—the breakdown of glucose to pyruvate. Glycolysis and gluconeogenesis utilize many, but not all, of the same enzymes; therefore, only one of these processes can occur in a tissue at any one time.
Although the two pathways act in a reciprocal manner and share enzymes, each also utilizes unique enzymes. It is these unique enzymes that are critical to the control of these two pathways.
Although the two pathways act in a reciprocal manner and share enzymes, each also utilizes unique enzymes. It is these unique enzymes that are critical to the control of these two pathways.
Gluconeogenesis occurs primarily in the liver. In this pathway, pyruvate is converted to oxaloacetate and then ultimately to glucose. The enzymes unique to gluconeogenesis are pyruvate carboxylase, fructose 1,6-bisphosphatase, and glucose 6-phosphatase. The pathway begins in the mitochondrion, where pyruvate is converted to oxaloacetate by pyruvate carboxylase, the first rate-limiting enzyme in the pathway. Pyruvate carboxylase is regulated by levels of acetyl-CoA (coenzyme A), which is required as a positive effector. Oxaloacetate is then reduced to malate by malate dehydrogenase and transported into the cytosol of the hepatocyte. There, malate is reoxidized into oxaloacetate. The oxaloacetate is then converted to phosphoenolpyruvate by phosphoenolpyruvate carboxykinase, another important enzyme in gluconeogenesis. Phosphoenolpyruvate is then converted to fructose 1,6-bisphosphate through a series of reactions catalyzed by enzymes shared with the glycolytic pathway.
Fructose 1,6-bisphosphate is converted to fructose 6-phosphate by another enzyme unique to gluconeogenesis, fructose 1,6-bisphosphatase. Next, fructose 6-phosphate is converted to glucose 6-phosphate by phosphohexose isomerase. Fructose 1,6-bisphosphatase is the second rate-limiting enzyme. It is induced by high levels of acetyl-CoA and suppressed by high levels of glucose 6-phosphate. Fructose 1,6-bisphosphatase is also regulated by fructose 2,6-bisphosphate. Fructose 2,6-bisphosphate is the regulator through which glucagon inhibits fructose 1,6-bisphosphatase. Glucose 6-phosphate is then dephosphorylated by glucose 6-phosphatase to glucose in the last rate-limiting step. The activity of this enzyme is suppressed by insulin.
Glycolysis occurs in cells throughout the body. The first step in the metabolism of glucose is entry of the glucose into the cytoplasm of the cell. Glucose uptake is largely insulin mediated, particularly in muscle and adipose tissue. Without insulin, cellular uptake and therefore glycolysis are greatly reduced. Once in the cytoplasm, glucose is phosphorylated by hexokinase (or glucokinase in the liver) into glucose 6-phosphate. This is the first rate-limiting step in glycolysis. Hexokinase is inhibited by its product, glucose 6-phosphate, and is induced by insulin.
Glucose 6-phosphate is converted to fructose 6-phosphate by phosphohexose isomerase. Then fructose 6-phosphate is converted to fructose 1,6-bisphosphate by phosphofructokinase-1. The activity of phosphofructokinase-1, the next rate-limiting enzyme, is regulated by glucagon through fructose 2,6-bisphosphate. Then, fructose 1,6-bisphosphate is converted through a series of reactions to phosphoenolpyruvate. In the last step, phosphoenolpyruvate is transformed by pyruvate kinase.
Glycogen synthesis and glycogenolysis occur primarily in liver and muscle. The key step in glycogenolysis is catalyzed by the enzyme glycogen phosphorylase a. Glycogen synthase is the key enzyme in glycogen synthesis. Glucagon and epinephrine act to inactivate glycogen synthase and activate glycogen phosphorylase, thereby inducing glycogenolysis and inhibiting glycogen synthesis.
DEHYDRATION AND HYPEROSMOLARITY
In both HHS and DKA, hyperglycemia results in an osmotic diuresis, which leads to dehydration. Hyperosmolarity develops, as the urine is relatively hypo-osmolar. Initially, the glycosuria causes an increase in the glomerular filtration rate. Once hypovolemia becomes significant, the glomerular filtration rate decreases and renal glucose losses may decrease as well. As renal clearance of glucose declines, hyperglycemia and hyperosmolarity worsen.
KETOGENESIS AND ACIDOSIS
Although the same mechanisms cause hyperglycemia and dehydration in both DKA and HHS, DKA is distinguished by ketogenesis (Fig. 53.3). Laffel (26) provides an excellent review of
ketone metabolism. In the adipocyte, hormone-sensitive lipase converts triglycerides into diglycerides, which are then converted into free fatty acids. Hormone-sensitive lipase is stimulated by counterregulatory hormones and inhibited by insulin.
ketone metabolism. In the adipocyte, hormone-sensitive lipase converts triglycerides into diglycerides, which are then converted into free fatty acids. Hormone-sensitive lipase is stimulated by counterregulatory hormones and inhibited by insulin.
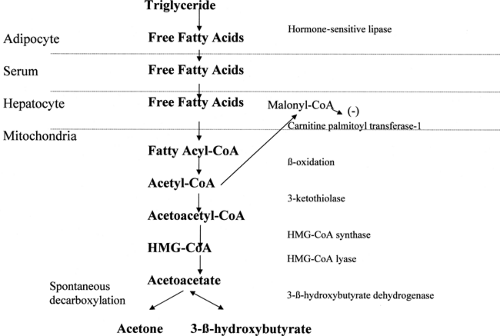 Figure 53.3. Glucose metabolism. HMG-CoA, hydroxymethylglutaryl–coenzyme A. |
Free fatty acids are converted in the cytoplasm of the hepatocyte to fatty acyl-CoA. Fatty acyl-CoA is transported into the mitochondria of the hepatocyte by the carnitine shuttle. Glucagon inhibits the enzyme acetyl-CoA carboxylase, which leads to reduced levels of malonyl-CoA. Malonyl-CoA normally inhibits carnitine palmitoyl transferase-1 (CPT-1), the rate-limiting step in the carnitine shuttle (26). Once in the mitochondrion, the fatty acyl-CoA undergoes β-oxidation to acetyl-CoA. Acetyl-CoA is then converted in the mitochondrion to acetoacetate. The rate-limiting step is HMG-CoA (hydroxymethylglutaryl-coenzyme A) synthase. Acetoacetate, in turn, is then either spontaneously decarboxylated into acetone or converted to 3-β-hydroxybutyrate by 3-βhydroxybutyrate dehydrogenase. Under normal circumstances, the ratio of 3-β-hydroxybutyrate to acetoacetate is about 1:1. Acetone is present in much lower quantities. In DKA, although levels of all three ketone bodies increase, the level of 3-β-hydroxybutyrate increases dramatically relative to that of acetoacetate, with ratios that may reach 10:1 (27).
Normally, ketone bodies increase insulin release from the pancreas; the insulin in turn suppresses ketogenesis. In the insulin-deficient state, the pancreatic β-cells are unable to respond, and ketogenesis proceeds unchecked.
Ketolysis occurs in the mitochondria of organs, which can use ketone bodies as an alternative energy source. Ketone bodies provide an important source of energy for the central nervous system during periods of starvation. However, skeletal muscle is probably the largest contributor to ketolysis. Acetoacetate is converted back to acetoacetyl-CoA by succinyl-CoA oxoacid transferase (SCOT). This is the rate-limiting step in ketolysis. Acetoacetyl-CoA is then converted to acetyl-CoA by methylacetoacetyl-CoA thiolase (MAT). Some ketones are also eliminated via the urine. Acetoacetate and 3-β-hydroxybutyrate are strong organic acids, which dissociate readily and account for the acidosis of DKA. Acetone does not dissociate and therefore does not worsen acidosis. It is excreted slowly via the lungs.
PRECIPITATING CAUSES OF DIABETIC KETOACIDOSIS AND HYPEROSMOLAR HYPERGLYCEMIC STATE
Infection remains the most common precipitating cause of DKA and HHS and accounts for 20% to 40% of all cases of DKA and HHS (28). The most common infections remain those of the urinary tract and lungs. Even though patients’ caloric intake is decreased during intercurrent illness, most of them need to increase their insulin dose and fail to do so. Elderly patients with diabetes often neglect the increases in polyuria and polydipsia that are associated with increasing glucose concentrations during periods of intercurrent illness and present for medical attention at a more advanced stage of their illness. Up to 20% of individuals with newly diagnosed cases of type 1 diabetes, and even some with cases of type 2 diabetes, present with DKA. Some subjects with type 2 diabetes who present with DKA as the first manifestation of the disease are able to stop insulin and take oral antidiabetic agents once blood sugars have stabilized. Other precipitating causes of DKA and HHS include cerebrovascular accidents, myocardial infarction, pancreatitis, and alcohol abuse. These causes account for about 10% of all cases of DKA and HHS. Omission of insulin remains a common cause of DKA in the adolescent population, many of whom utilize this as an opportunity to control their weight (29). This is also a common cause of DKA in African Americans (30). Drugs also may precipitate DKA and HHS, with most frequent offenders being steroids, sympathomimetic agents (dobutamine and terbutaline), and thiazides. Recently, newer antipsychotic drugs (e.g., clozapine, olanzapine, and risperidone) have been associated with the development of hyperglycemia, and patients have presented with DKA as the first manifestation of their diabetes (31,32,33,34,35,36,37). Therapy with interferon-α and ribavirin has been associated with the development of DKA in a patient with hepatitis C (38). DKA has been reported in association with the use of protease inhibitors and pentamidine therapy, and recreational use of cocaine and “ecstasy” has also been reported to precipitate DKA (39,40,41,42,43). In some patients, no obvious precipitating cause can be found.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


