Pathophysiology and Treatment of Lipid Disorders in Diabetes
Barbara V. Howard
Wm. James Howard
An understanding of lipoprotein metabolism and how it influences diabetes is of particular importance because of the association of lipoproteins with cardiovascular disease, the leading cause of death among people with diabetes (1). Abnormalities in lipoproteins are very common in both type 1 and type 2 diabetes. Although alterations in lipoproteins appear to be an intrinsic part of these disorders, such alterations also are induced by diabetes-associated complications such as obesity and renal disease and are sometimes exacerbated by therapeutic regimens associated with the management of diabetic patients. The National Cholesterol Education Program (2) and the American Diabetes Association (ADA) (3) have focused attention on the necessity of managing lipid disorders. In diagnosing and treating lipid abnormalities in diabetes, particular consideration must be given to diabetes-specific targets, the relationship between glycemic control and lipoproteins, and the potential for a different response to lipid-lowering agents by individuals with diabetes. This chapter will review the basics of lipoprotein composition and metabolism, the alterations in lipoprotein composition and metabolism in diabetes, and therapeutic approaches to the management of lipid disorders in patients with diabetes.
LIPOPROTEIN METABOLISM
Structure and Classification
Lipoproteins are microemulsions composed of lipids (cholesterol, cholesteryl ester, triglyceride, and phospholipid) and proteins (apoproteins). Their function is to transport non-water-soluble cholesterol and triglycerides in plasma. Lipoproteins are spherical particles containing a central core of nonpolar lipids (primarily triglycerides and cholesteryl ester) and a surface monolayer of phospholipids and apoproteins. Free cholesterol is present primarily in the surface monolayer. [For a detailed review of lipoprotein structure and metabolism, the reader is referred to references (4) through (16)].
Lipoproteins have been classified on the basis of their densities during ultracentrifugation (Table 33.1). Chylomicrons, particles that are primarily triglyceride bearing, are produced by the intestine after exogenous fat undergoes digestion. Very-low-density lipoproteins (VLDLs), also triglyceride-bearing lipoproteins, are secreted by the liver and carry endogenously produced triglyceride. Intermediate-density lipoproteins (IDLs) represent remnants of the metabolism of triglyceride-rich lipoproteins and also can be intermediates in the conversion of VLDLs to low-density lipoproteins (LDLs) (8,13,14,15,16). LDLs are the major cholesterol-bearing lipoproteins and are those most strongly related to the occurrence of cardiovascular disease. Lp(a) is a subclass of the LDL fraction that consists of LDL complexed to a large glycoprotein resembling plasminogen; this complex has also been associated with atherosclerosis (17). High-density lipoproteins (HDLs) are the smallest and densest of the lipoproteins. Although HDLs also transport substantial amounts of cholesterol, they are negatively associated with cardiovascular disease.
TABLE 33.1. Human Plasma Lipoproteins | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The metabolism and production of all lipoproteins are controlled by the apoproteins contained within the complex, by enzymes and proteins that moderate lipoprotein metabolism, and by specific cellular receptors that direct their interaction with cells. All known apoproteins (Table 33.2) have been sequenced and their genes localized (18,19). Most are hydrophobic proteins and serve as ligands for specific receptors involved in the metabolism of the various lipoproteins and as cofactors for enzymatic activities involved in lipoprotein metabolism. Several other proteins and enzymes play key roles in plasma lipoprotein transport, including lipoprotein lipase (8,9,16,20,21) and hepatic lipase (12,22), which catalyze the delipidation of triglyceride-rich particles; lecithin-cholesterol acyltransferase (LCAT) (23), which is responsible for the synthesis of virtually all cholesteryl esters in plasma lipoproteins; and cholesteryl ester transfer protein (CETP) (24,25), which facilitates the transfer of cholesteryl ester and triglycerides between lipoproteins during their metabolism. A number of cell receptors (Table 33.3) govern lipoprotein binding and uptake and lipid flux; they are described in the following sections on metabolism.
TABLE 33.2. Major Apoproteins Associated with Plasma Lipoproteins | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 33.3. Cell Receptors Involved in Lipoprotein Metabolism | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Formation and Metabolism of Chylomicrons
Chylomicrons are responsible for the transport of dietary triglycerides and cholesterol. Dietary triglycerides are hydrolyzed in the gut, releasing monoglycerides and fatty acids that are then reesterified to form triglycerides in the intestinal mucosal cell (Fig. 33.1) (5,7,8,10,11,16). These triglycerides are assembled with newly absorbed cholesterol, apoprotein (apo) B48, and the A apoproteins. Upon secretion from the enterocyte, these assembled particles enter the lymphatic circulation and then the bloodstream, where they acquire C apoproteins and apoE by transfer from HDL. As chylomicrons enter the plasma, the triglycerides are rapidly hydrolyzed by the enzyme lipoprotein lipase (LPL), which resides on the surface of capillary endothelial cells. LPL is synthesized primarily in adipose tissue and striated muscle (8,10,16,20,21). It is secreted and transported to the endothelial surface, where it acts on triglyceride-rich particles. Its action requires the presence of apoCII on the
surface of the lipoprotein, whereas apoCIII inhibits LPL. LPL is induced in adipose tissue by insulin (26). The liberated free fatty acids are available for oxidative needs of peripheral cells, and excess free fatty acids are stored primarily in triglycerides in adipose tissue to serve as a future source of free fatty acids.
surface of the lipoprotein, whereas apoCIII inhibits LPL. LPL is induced in adipose tissue by insulin (26). The liberated free fatty acids are available for oxidative needs of peripheral cells, and excess free fatty acids are stored primarily in triglycerides in adipose tissue to serve as a future source of free fatty acids.
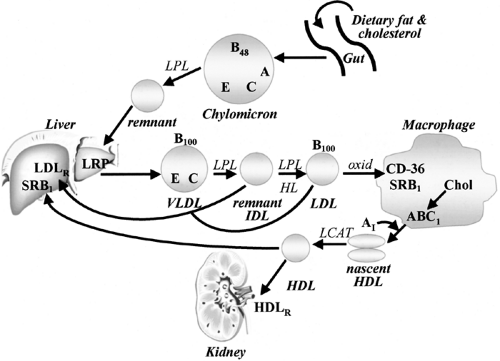 Figure 33.1. Schematic of lipoprotein metabolism. LDLR, low-density lipoprotein receptor; SRB1, scavenger receptor class B, type 1; LRP, LDL receptor-like protein; LPL, lipoprotein lipase; VLDL, very-low-density lipoproteins; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; HL, hepatic lipase; oxid, oxidation; CD-36, conjugated diene lipid hydroperoxide; Chol, cholesterol; ABCl, ATP binding cassette 1; LCAT, lecithin-cholesterol acyltransferase; HDLR, HDL receptor; B48, E, CII, B100, AI, apoproteins (apo). |
As triglyceride is depleted from the chylomicrons, phospholipids and A and C apoproteins are transferred to HDL. The residual chylomicron particle, which has lost 80% to 90% of its triglyceride and is now relatively cholesterol-enriched, is called a chylomicron remnant. These remnants are believed to be cleared by the liver via LDL receptor-like protein (LRP), a protein that resembles the apo B/E receptor but probably recognizes apoB48 (27). The remnants thus enter lysosomes in the liver, from which cholesterol can enter metabolic pathways in the hepatocytes, including excretion into the bile. The remaining triglyceride enters the hepatic triglyceride stores.
Very-Low-Density Lipoproteins
VLDLs are synthesized in the endoplasmic reticulum of hepatocytes and are composed of endogenous triglyceride derived from plasma free fatty acids, from chylomicron remnants, and from de novo lipogenesis (Fig. 33.1) (5,7,8,11,14,15). Post-translational regulation of VLDL assembly by glucose, fatty acids, and insulin is the major determinant of secretion (28). The concentration of circulating free fatty acids governs the rate of triglyceride esterification in the liver, and glucose—especially when glycogen synthesis is impeded—may stimulate de novo production of free fatty acids and thus triglycerides. Triglyceride transfer protein in the hepatocyte regulates the transport and assembly of triglyceride with apoB (28,29). Insulin is required for VLDL production, both for apoprotein synthesis and for its role in regulating several enzymes involved in lipogenesis (30). On the other hand, increases in insulin have been shown to inhibit VLDL secretion in hepatocytes by phosphorylating apoB, which impedes the assembly of apoB with lipids (28,31), and increases in insulin have been shown to inhibit VLDL production in vivo in human studies (32).
Nascent VLDLs, as secreted into the circulation, contain apoB100 and small amounts of apoC and apoE. After VLDLs enter the circulation, they are metabolized in the same manner as chylomicrons by the enzyme LPL, with the fatty acids that are liberated following the same fate as those liberated from chylomicrons. After secretion, VLDLs acquire more C and E apoproteins by transfer from HDL. In addition, free cholesterol is progressively exchanged to HDL, where it is esterified and the cholesteryl ester is returned to VLDL. As VLDLs become progressively depleted of triglyceride, a portion of the surface, including cholesterol, apolipoproteins C and E, and phospholipids, is removed and contributes to nascent HDL particles (33). The enzyme hepatic lipase also plays a role in the metabolism of smaller VLDL particles during the latter stages of the VLDL catabolic cascade (22). The smaller remnants of VLDL are triglyceride-depleted, cholesterol-rich particles, some of which are isolated in the IDL compartment, although some remain in the VLDL compartment. These remnant particles are cleared from the circulation primarily by receptors in the liver. These receptors include both the B/E receptor (see below) and possibly LRP (27), which acts on chylomicron remnants. A portion of VLDL remnants is further metabolized, possibly within the intracellular spaces of the liver, by a process involving hepatic lipase, to form LDL. During this process, the remainder of apoproteins other than apoE is lost.
The mechanisms that determine which and how many VLDL particles are converted to LDL are not clearly understood. VLDL particles are believed to be secreted in a spectrum of sizes with various degrees of triglyceride enrichment. The larger VLDL particles appear to be more rapidly cleared and less likely to be converted to LDL (34). On the other hand, smaller VLDL particles that are richer in cholesterol may be preferentially converted to LDL. When apoE is missing or defective (as in type III hyperlipidemia) (14), clearance of chylomicron remnants and VLDL remnants is much slower than normal. These remnants accumulate in plasma and are not readily converted to LDL.
Low-Density Lipoproteins
As indicated above, LDLs are products of the metabolism of VLDL (Fig. 33.1) (4,5,7,8). The only apolipoprotein in LDL is apoB, and only one molecule of apoB is present per particle of LDL. Clearance of LDL is mediated by a specific receptor (the B/E receptor) present on the surface of both liver and peripheral cells (4). Once it is bound to the receptor, the lipoprotein is internalized by an endocytotic process (Fig. 33.2). The vesicle then fuses with a lysosome, where enzymes degrade the apoB and hydrolyze the cholesteryl ester to free cholesterol. Triglycerides and phospholipids may also be hydrolyzed. The influx of free cholesterol from LDL sets into motion a cascade of regulatory events aimed at controlling the cholesterol content of the cell. Esterification of cholesterol is stimulated by activation of acyl CoA cholesterol acyltransferase (ACAT). Simultaneously, de novo cholesterol production is inhibited by the inhibition of 3-hydroxy-3-methyl-glutaryl (HMG)-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis. Finally, accumulation of intracellular cholesterol limits the further uptake of cholesterol-rich lipoproteins by inhibiting synthesis of the B/E receptor.
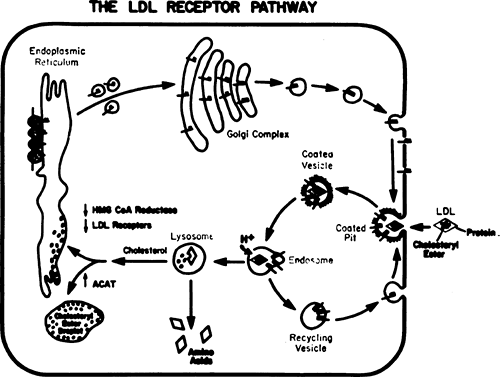 Figure 33.2. Binding and cellular metabolism of low-density lipoprotein (LDL). HMG, 3-hydroxy-3-methyl-glutaryl; ACAT, acyl CoA cholesterol acyltransferase. (From Brown MS, Goldstein JL. The LDL receptor and HMG-CoA reductase—two membrane molecules that regulate cholesterol homeostasis. Curr Top Cell Regul 1985;26:3–15, with permission.) |
LDL may also be removed by clearance mechanisms mediated not by the B/E receptor but by phagocytic cells; such mechanisms include uptake by both nonspecific endocytosis and via the scavenger receptors—classes A (SRA) and B type 1 (SRB1, known as CLA1 in humans), and conjugated diene lipid hydroperoxide 36 (CD-36) receptors, which recognize altered (oxidized or glycated) LDL (35,36,37,38,39). This process is thought to be responsible for cholesterol deposition in macrophages of the vessel wall.
LDLs represent a spectrum of particles varying in density from larger cholesterol-rich particles (phenotype A) to smaller cholesterol-poor particles (phenotype B) (40,41). The latter usually occur when levels of triglyceride-rich lipoproteins are elevated as a result of CETP-mediated exchange between triglyceride and the cholesteryl ester from LDL followed by lipolysis of LDL triglyceride (42). These smaller, denser LDL particles are more susceptible to modification by oxidation (and glycation, see below), after which they are recognized and taken up by macrophages.
High-Density Lipoproteins
HDLs also are represented by a spectrum of particles of various sizes and densities (5,7,13). HDLs are secreted by the hepatocyte as small, cholesterol-poor/protein-rich particles that contain the A apoproteins as well as apoE. Small (nascent) HDLs also are produced by the intestine (Fig. 33.1). Some HDL particles contain both apoAI and apoAII, whereas others contain only apoA1. In the plasma, surface components of triglyceride-rich lipoproteins are transferred to HDL during lipolysis. In addition,
HDL particles are the sites of synthesis of cholesteryl ester from free cholesterol and lecithin through the enzyme LCAT, which circulates in association with HDL particles. HDLs participate in the catabolism of triglyceride-rich lipoproteins, serving as a source for cholesteryl esters and the C and E apoproteins and ultimately as the receptacle for surface components. During the lipolytic process, the size of the HDL particle increases. Larger particles are referred to as HDL2 and the smaller HDL precursors are known as HDL3.
HDL particles are the sites of synthesis of cholesteryl ester from free cholesterol and lecithin through the enzyme LCAT, which circulates in association with HDL particles. HDLs participate in the catabolism of triglyceride-rich lipoproteins, serving as a source for cholesteryl esters and the C and E apoproteins and ultimately as the receptacle for surface components. During the lipolytic process, the size of the HDL particle increases. Larger particles are referred to as HDL2 and the smaller HDL precursors are known as HDL3.
HDLs, along with LCAT and CETP (Table 33.4), are important in the flux of cholesterol from cells to plasma lipoproteins and the liver (13,24,25,43,44). The ATP binding cassette 1 (ABC1) receptor, which is present on macrophages and other peripheral cells, facilitates the transfer of cellular cholesterol to apoA1, which is free in plasma or on nascent HDL particles. After conversion to cholesteryl ester by LCAT, the cholesteryl ester becomes trapped within the nonpolar center of HDL; this cholesteryl ester may circulate in HDL or be transferred to triglyceride-rich particles. The SRB1 receptor may also mediate the removal of cholesterol and cholesteryl ester from peripheral cells or macrophages to HDL through a process of selective lipid transfer. This receptor mediates the transfer of cholesterol and cholesteryl ester from cells to HDL and from larger HDLs to the liver. This process represents a pathway for “reverse cholesterol transport,” by which cholesterol from peripheral cells can be removed and transported to the liver for excretion in bile.
TABLE 33.4. Important Plasma Regulators of Lipoprotein Metabolism | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
The mechanisms that control clearance of HDL are not well understood. Hepatic lipase may hydrolyze HDL phospholipids, which in turn may promote net transfer of cholesterol from the surface of HDL to the liver. A specific HDL receptor (HDLR) on the kidney catalyzes specific uptake and degradation of HDL particles (45). This receptor also provides cholesterol to adrenal cells for steroidogenesis.
LIPOPROTEIN ALTERATIONS IN TYPE 2 DIABETES
The following summarizes commonly observed lipoprotein changes and their possible metabolic determinants in individuals with type 2 diabetes (Table 33.5) (for additional details, see references 30,46,47,48,49,50,51,52,53,54,55).
TABLE 33.5. Lipoprotein Alterations in Type 2 Diabetes | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Triglycerides and Very-Low-Density Lipoproteins
The most common alteration of lipoproteins in type 2 diabetes is hypertriglyceridemia caused by an elevation in VLDL concentrations. In clinical descriptions of diabetic hypertriglyceridemia, an emphasis is often placed on individuals with extremely high levels of plasma and VLDL triglycerides. It is clear, however, from population-based studies (53,56) that type 2 diabetes generally is associated with only a 50% to 100% elevation in the plasma levels of total and VLDL triglycerides. Thus, it is likely that subjects with type 2 diabetes who have concentrations of total triglycerides greater than 350 to 400 mg/dL also have genetic defects in lipoprotein metabolism, the expression of which may be exacerbated by hyperglycemia (57) (see below).
Metabolic Determinants
One of the determinants of diabetic hypertriglyceridemia is the overproduction of VLDL triglyceride (58,59,60), which is most likely due to the increased flow of substrates, particularly glucose and free fatty acids, to the liver. In addition, individuals with type 2 diabetes appear to have a defect in clearance of VLDL triglyceride that parallels the degree of hyperglycemia (58,59,60,61). Studies to date suggest that LPL activity is decreased in individuals with type 2 diabetes, especially those with moderate to severe hyperglycemia who exhibit both insulin deficiency and insulin resistance (62). However, in vivo clearance defects have not consistently been observed in patients with type 2 diabetes, especially in those with greatly elevated triglyceride levels.
The metabolism of VLDL apoB may also be altered in type 2 diabetes. Subjects with type 2 diabetes have a decreased fractional catabolic rate for VLDL apoB similar to that for VLDL triglyceride (60). Overproduction of VLDL apoB also occurs in
type 2 diabetes, and it has been suggested that this overproduction is further increased by obesity (60). Although obese diabetic subjects have a higher VLDL-B production than do lean individuals, VLDL-B production may already be maximally stimulated in obese nondiabetic individuals (60). Thus, the extent of overproduction of VLDL triglyceride may be greater than that of apoB in type 2 diabetes, a situation that results in the production of larger triglyceride-rich VLDL particles.
type 2 diabetes, and it has been suggested that this overproduction is further increased by obesity (60). Although obese diabetic subjects have a higher VLDL-B production than do lean individuals, VLDL-B production may already be maximally stimulated in obese nondiabetic individuals (60). Thus, the extent of overproduction of VLDL triglyceride may be greater than that of apoB in type 2 diabetes, a situation that results in the production of larger triglyceride-rich VLDL particles.
The alterations in VLDL metabolism in type 2 diabetes are related in part to insulin resistance. Several studies have shown correlations between VLDL concentrations and measures of insulin resistance (30,52). Hyperinsulinemia and the central obesity that typically accompanies insulin resistance also are thought to lead to overproduction and impaired catabolism of VLDL. Metabolic alterations in insulin resistance that lead to VLDL overproduction include (a) increased free fatty acid and glucose levels, which regulate VLDL output from the liver, and (b) elevated triglyceride levels in the liver, which inhibit apoB degradation and result in increased assembly and secretion of VLDL. In addition, LPL levels are reduced in insulin resistance, which interferes with the normal lipoprotein metabolic cascade and results in decreased clearance of VLDL.
Triglyceride elevations in type 2 diabetes may also be due to delayed clearance of postprandial particles (63). Mechanisms of delayed clearance of chylomicrons are similar to those discussed for VLDL clearance.
Very-Low-Density Lipoprotein Composition
In addition to increases in the amount of VLDL, changes in VLDL composition in type 2 diabetes may reflect or be the cause of alterations in VLDL metabolism. Several studies suggest that individuals with diabetes, especially those with severe hyperglycemia, may have larger triglyceride-rich VLDL (60,64). This increased ratio of triglyceride to apoB may be a reflection of a disproportionate influence of type 2 diabetes on VLDL triglyceride production (see above). Subfractions of VLDL have been found to be enriched in the proportion of cholesterol-rich particles (65). These compositional changes may have implications for the increased propensity for atherosclerosis among individuals with type 2 diabetes, because cholesterol-enriched VLDL may be atherogenic. Changes in the distribution of apoE would have important implications for VLDL metabolism in type 2 diabetes because apoE influences the affinity of binding to receptors. An increased proportion of apoE in the VLDL of type 2 diabetes has been reported (66). Although differences in distribution of apoE phenotypes in diabetes have not been demonstrated, apoE sialation has been reported to be higher in diabetic than nondiabetic individuals, a change that may impair binding to the B/E receptor (67).
Additional evidence for abnormal VLDL in type 2 diabetes is that VLDLs from persons with type 2 diabetes have altered metabolic properties in vitro. VLDL isolated from normotriglyceridemic patients with type 2 diabetes produced a greater cellular accumulation of lipids in mouse peritoneal macrophages than did VLDL isolated from either normotriglyceridemic or hypertriglyceridemic nondiabetic control subjects (68). Thus, altered VLDL composition may contribute to metabolic abnormalities as well as to the atherosclerotic propensity of the VLDL particles.
Remnant particles from delayed chylomicron clearance may also be present in the VLDL fraction; they are subject to the same compositional alterations discussed for VLDL.
Low-Density Lipoprotein Cholesterol
Studies examining plasma concentrations of total and LDL cholesterol in type 2 diabetes vary by population, with some showing higher and some showing lower levels in type 2 diabetes than in control subjects. Data from the National Health and Nutrition Examination Survey (NHANES II) indicate that levels of LDL cholesterol in whites with diabetes are higher than for blacks with diabetes, after adjustment for relevant covariates (56). Lower LDL concentrations are also seen in American Indians with diabetes (53). It should be pointed out that in most population studies, the density ranges chosen for quantitation of LDL (1.006 to 1.063) result in the inclusion of the IDL fraction. It is possible that the increase in LDL in type 2 diabetes is the result of an increase in this IDL fraction.
METABOLISM
In individuals with type 2 diabetes and relatively severe hyperglycemia, the clearance rate for LDL apoB is reduced (60,69). Mildly hyperglycemic individuals with type 2 diabetes may have increased LDL production as well (69). Because LDL binding is stimulated by insulin (70), defects in LDL clearance in type 2 diabetes may be due to insulin resistance or relative insulin deficiency. This possibility is supported by the observation that clearance of LDL in type 2 diabetes is positively related to plasma insulin levels and to the insulin response from oral glucose challenge (69). Direct removal of VLDL apoB is also increased in individuals with type 2 diabetes and large, triglyceride-rich VLDL (60). Thus, the concentrations of LDL in type 2 diabetes may be influenced by two opposing phenomena. On one hand, decreased clearance in type 2 diabetes may lead to increased LDL; on the other hand, increased direct removal tends to lower production. The resultant concentration may thus be dependent on the relative magnitude of these two processes. Nevertheless, these alterations in the flux of both VLDL remnants and LDL particles, coupled with the changes in LDL composition, indicate that LDL in individuals with type 2 diabetes has significant atherogenic potential.
COMPOSITION
The composition of LDL in type 2 diabetes is altered, and these changes also may contribute significantly to abnormal metabolism and atherosclerosis. An increase in the proportion of small, dense, triglyceride-enriched LDL has consistently been observed (64,71,72,73). LDL particles from individuals with diabetes have a decreased ability to bind to receptors, and this decrease in binding is inversely related to the size and ratio of triglyceride to protein in LDL (74). LDL in diabetic individuals has been shown to be more rapidly oxidized (75). This may be in part because of the increased oxidative susceptibility of small, dense LDL particles, which are prevalent in diabetic individuals. Oxidized LDL particles are believed to play a major role in stimulating the atherosclerotic process because of their recognition by macrophage receptors (Table 33.3).
Increased plasma triglyceride levels, low HDL levels, and small, dense LDLs usually occur together in a lipoprotein pattern often referred to as atherogenic dyslipidemia (76). This abnormal pattern occurs in insulin resistance, is exacerbated in diabetes (52,77), and is derived in part from alterations in apoB metabolism because triglyceride-rich VLDLs are the precursors of denser LDL particles (78). Small, dense LDLs are slowly catabolized because they do not bind well to the B/E receptor.
Nonenzymatic glycation (or glycosylation) of apoB also may influence LDL metabolism in diabetes. Small, dense LDLs are more rapidly glycated. The extent of glycation of LDL in individuals with type 2 diabetes who have moderate hyperglycemia is approximately 2% to 5% (79), and this degree of glycation of lysine residues has been shown to decrease LDL catabolism in vivo by 5% to 25% (80). Glycated LDLs also appear to exhibit altered interactions with endothelial cells, stimulate cytokine production,
and enhance cholesteryl ester synthesis in human macrophages (81). Moreover, glycated LDLs are more readily oxidized. Thus, the glycation of LDL may represent an important mechanism by which atherogenesis is increased in type 2 diabetes. Together, glycation and oxidation render LDLs more immunogenic; the formation of antibody-antigen complexes stimulates macrophage accumulation and further foam cell formation (82).
and enhance cholesteryl ester synthesis in human macrophages (81). Moreover, glycated LDLs are more readily oxidized. Thus, the glycation of LDL may represent an important mechanism by which atherogenesis is increased in type 2 diabetes. Together, glycation and oxidation render LDLs more immunogenic; the formation of antibody-antigen complexes stimulates macrophage accumulation and further foam cell formation (82).
Finally, a pattern of abnormal cholesterol transport and transfer in the plasma has been shown in patients with type 2 diabetes. The transfer of LCAT-synthesized cholesteryl esters to VLDL and LDL is inhibited, with a concomitant increase in their transfer to HDL; this abnormal metabolic pattern is reversed by insulin therapy (83). The block in cholesteryl ester transfer activity in patients with type 2 diabetes is correlated with an increase in free cholesterol content of both LDL and VLDL. Therefore, in type 2 diabetes, this abnormal cholesteryl ester transfer may be related to an increased risk for atherosclerosis.
High-Density Lipoprotein Cholesterol
Almost as common as the observation of increased VLDL concentrations in type 2 diabetes is the finding of decreased concentrations of HDL cholesterol in individuals with type 2 diabetes.
METABOLISM
Individuals with type 2 diabetes have an increased rate of HDL clearance, as measured by apoAI and apoAII kinetics (84,85,86). Significant correlations have been found between HDL clearance and plasma concentrations of HDL cholesterol and apoA1, and the increase in HDL clearance was directly related to plasma glucose levels. The finding of increased HDL clearance is consistent with lower VLDL clearance and lower LPL activity. Because HDL concentrations, especially of larger HDLs, increase during the lipolytic process, the decreases in LPL activity and impaired VLDL catabolism have been shown to be correlated with decreases in HDL concentrations in patients with type 2 diabetes. Elevated hepatic lipase activity also may contribute to the decrease in HDL concentrations in type 2 diabetes, because this enzyme also plays a key role in the metabolism of HDL. The changes in lipoprotein and hepatic lipases may act in concert to decrease HDL levels in type 2 diabetes.
COMPOSITION
There are several indications that the composition of HDLs in type 2 diabetes may be altered (52). These differences may be in part a reflection of alterations in the delipidation cascade. Decreased HDL concentrations in type 2 diabetes are reflected mostly in decreases in larger particles. As with LDL, in type 2 diabetes an increased proportion of triglyceride in HDL has been observed. These compositional changes appear to be related to the activity of adipose tissue LPL, because LPL deficiency may be a factor responsible for the altered distribution of HDL particles in untreated type 2 diabetes. Nonenzymatic glycation of HDL appears to interfere with HDL receptor binding (87). Thus, glycation of HDL may also play a role in the lower levels of HDL observed in diabetes. Finally, abnormalities in HDL composition have been noted even in individuals with optimal glycemic control (88). All of these alterations in HDL composition may impair the role of HDL in reverse cholesterol transport.
As stated above, the decreases in HDL concentration and changes in HDL composition are part of the dyslipidemia of insulin resistance. Changes in HDL metabolism in insulin resistance include (a) impaired VLDL lipolysis, which depletes HDL by impeding the transfer of apoproteins and cholesterol ester from triglyceride-rich lipoproteins to the HDL compartment; (b) increased activity of hepatic lipase, which facilitates HDL clearance; and (c) alterations in hepatic function, which inhibit production of apoAI (the main apoprotein of HDL) and/or hepatic secretion of nascent HDL (52).
Significant negative relationships between plasma concentrations of insulin and HDL have been observed in subjects with type 2 diabetes as well as a negative relationship between insulin resistance and HDL cholesterol that is independent of VLDL concentrations (53,63). These observations indicate that insulin or insulin resistance influences the concentration or composition of HDL in some way.
LIPOPROTEINS IN TYPE 1 DIABETES
The consideration of lipoprotein metabolism in type 1 diabetes is influenced by the requirement for insulin therapy. Thus, a spectrum of situations is possible, from the insulin-deficient ketoacidotic state with greatly elevated glucose, free fatty acids, ketones, and lipolytic hormones such as glucagon and epinephrine, to that seen when continuous insulin therapy is administered, in which an excess of insulin in peripheral plasma is found and glucose and fatty acid levels are close to normal (Fig. 33.3) (47,48,49,50,51,52). In the following sections, an attempt will be made to differentiate between these various degrees of control.
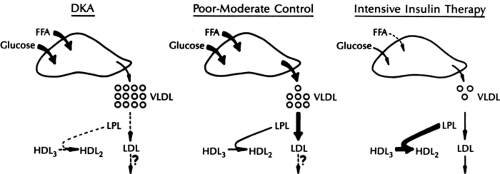 Figure 33.3. Spectrum of lipoprotein changes in type 1 diabetes with various degrees of control. DKA, diabetic ketoacidosis. See legend to Figure 33.1 for other abbreviations. (From Howard BV. Lipoprotein metabolism in diabetes mellitus. J Lipid Res 1987;28:613–628, with permission.) |
Very-Low-Density Lipoproteins
Extreme elevations in VLDL levels have been recognized as being a common occurrence in diabetic ketoacidosis, the stage at which insulin concentrations are minimal (89). On the other hand, VLDL levels may not be elevated in individuals with type 1 diabetes who are receiving adequate therapy. It is now well established that elevations in VLDL triglycerides in type 1 diabetes are often correlated with the degree of diabetic control (90,91).
METABOLISM
In people with untreated type 1 diabetes, the fractional catabolic rate for endogenous triglyceride is decreased (92), as is the clearance rate for exogenous triglyceride (93). Thus, when insulin deficiency is extreme, clearance is impaired because the activity of LPL is dependent on insulin. In the early stages of insulin deficiency, production of VLDL is increased, probably because of the increase in mobilization of free fatty acids. This enhanced hepatic secretion of VLDL falls off in the later stages of ketoacidosis because of the decrease in hepatic protein synthesis secondary to the insulin deficiency. In poorly controlled but nonketotic patients with type 1 diabetes, both overproduction and decreased clearance are observed (94). Kinetic studies of VLDL triglyceride in subjects with type 1 diabetes with adequate conventional insulin therapy showed that production and fractional catabolic rates of VLDL triglycerides were normal when compared with rates in weight- and age-matched controls (95). Continuous subcutaneous insulin infusion produced a significant decline in VLDL triglyceride production to levels below those observed in nondiabetic subjects. There was no change in the mean fractional catabolic rate for VLDL triglyceride after insulin infusion. A similar decrease in plasma VLDL triglyceride production in type 1 diabetes was observed after treatment with the artificial β-cell (96). These latter studies indicate that rigorous insulin therapy can decrease rates of VLDL production and produce even subnormal levels of VLDL triglyceride.
During severe ketoacidosis when there is a marked insulin deficiency, hypertriglyceridemia is caused primarily by a deficiency in LPL activity, and overproduction of triglycerides may not occur despite elevated levels of free fatty acids. As insulin therapy is instituted, the situation changes. With moderate control (i.e., when insulin administration is suboptimal) there is both an overproduction of VLDL because of an increase in the level of free fatty acids and some deficiency in VLDL clearance because of the continued limitation in LPL. As stringent control is achieved with the administration of large amounts of peripheral insulin, VLDL clearance normalizes. VLDL production rates may fall to subnormal levels because excess insulin may suppress hepatic VLDL formation. Individuals with type 1 diabetes in this situation will have normal or even low-normal levels of circulating VLDL triglycerides.
Low-Density Lipoproteins
LDL concentration appears to vary directly with the extent of hyperglycemia. LDL levels are increased in poorly controlled type 1 diabetes. However, in many individuals with type 1 diabetes, LDL concentrations are not different from those of age- and weight-matched controls, and some type 1 diabetic subjects receiving insulin by means of a pump exhibit LDL concentrations considerably below those of controls.
METABOLISM
Fractional catabolic rates for LDL in subjects receiving conventional therapy or who have received 3 weeks of continuous subcutaneous insulin infusion are similar to those for nondiabetic controls. Improvement in glycemic control decreases LDL apoB production to levels below those of nondiabetic subjects. In uncontrolled type 1 diabetes, LDL fractional clearance is probably decreased because insulin appears to potentiate LDL binding to its receptor. With increased control, LDL metabolism may return to normal (97). Further, insulin deficiency may lead to overproduction of LDL in response to an increased influx of VLDL or its precursor or to impaired removal of VLDL remnants by the liver. Abnormalities in the VLDL particle also may influence conversion to LDL.
COMPOSITION
Individuals with type 1 diabetes may also have a preponderance of small, dense particles for the same reasons as those described for type 2 diabetes. LDLs isolated from patients with poorly controlled type 1 diabetes are taken up and degraded by fibroblasts at a lower rate than LDLs isolated from healthy subjects; when glucose concentrations are lowered by insulin therapy, the binding properties of LDLs return to normal (98). Glycated LDL and defects in cholesteryl ester transfer may be found in type 1, as well as in type 2, diabetes, although no studies on glycation in type 1 diabetes have yet been performed. Exposure of cells to lipoprotein-deficient serum obtained from patients with poorly controlled type 1 diabetes enhances the efficiency of LDL binding (99). It has been postulated that in type 1 diabetes, membrane changes may be induced that alter LDL binding.
High-Density Lipoproteins
It has been suggested that concentrations of HDL may be low in patients with untreated, insulin-deficient diabetes. Response of HDL to insulin therapy is slower than that of VLDL, but HDL increases with the degree of glycemic control. In several studies, HDL concentrations are similar or higher in patients with type 1 diabetes than in age-, sex-, and weight-matched controls. HDL levels may also be higher in those who are receiving insulin-pump therapy (91,92,93,94,95,96,97).
METABOLISM
One factor responsible for the decrease in HDL in patients with poorly controlled type 1 diabetes is low LPL activity. The reduced activity impairs lipolysis of VLDL and subsequently slows formation of HDL particles (62). Levels of both HDL cholesterol and phospholipids in type 1 diabetes have been shown to correlate positively with LPL activity; thus, greatly increased catabolism of triglyceride-rich lipoproteins in the presence of excess insulin might augment the HDL compartment. An inverse correlation has been observed between HDL and hepatic lipase activity in the plasma of type 1 diabetic subjects. Hepatic lipase activity may be lower in well-controlled type 1 diabetes and associated with a high ratio of HDL2 to HDL3; thus, the role of this enzyme in regulating HDL levels and the possible influence of insulin on this process are similar to those postulated for type 2 diabetes (100).
COMPOSITION
HDL elevations in well-controlled type 1 diabetes are generally due to an increase in larger particles. These observations confirm the hypothesis that the action of insulin, whether it occurs through the activity of LPL or via some other process, hastens the transfer of material to the HDL2 compartment. Data indicate that alterations in HDL composition may be found in type 1 diabetes much as they are in type 2 diabetes. Lower concentrations of HDL are associated with HDL particles that are smaller and richer in triglycerides. Abnormalities in both triglyceride and apoAI content have been observed in the HDL of individuals
with type 1 diabetes (101), and apoAI and apoAII content may also be decreased. ApoA has also been reported to be glycated in patients with type 1 diabetes (102).
with type 1 diabetes (101), and apoAI and apoAII content may also be decreased. ApoA has also been reported to be glycated in patients with type 1 diabetes (102).
RATIONALE FOR THERAPY FOR LIPID ABNORMALITIES IN DIABETES
Atherosclerotic macrovascular disease is the leading cause of morbidity and mortality in patients with diabetes mellitus. Both men and women with diabetes have a significantly increased risk of myocardial infarction (MI) (103,104,105), stroke, and peripheral gangrene (104,105). The risk for developing coronary heart disease (CHD) begins prior to the development of type 2 diabetes. By the time the diagnosis of type 2 diabetes is made, more than half of all diabetic individuals already have clinical CHD (106). Haffner et al. (107) compared this phenomenon to a clock that begins ticking with the onset of insulin resistance and accelerates with the development of hyperglycemia. In a Finnish cohort of patients with type 2 diabetes, Haffner and colleagues (108) demonstrated that diabetic patients without previous MI have as high a risk for a first MI as do nondiabetic patients with a history of MI for having a second infarction. In addition, patients with diabetes have an increased rate of MI-associated prehospital mortality (105,109), as well as increased morbidity and mortality during and after hospitalization (110,111,112). This is particularly true for women with diabetes (53,113) These data provide a strong rationale for treating cardiovascular risk factors in diabetic patients as aggressively as in nondiabetic patients with clinical CHD. Thus, diabetes confers a risk that is equivalent to that of known CHD. The ADA and the American Heart Association consider type 2 diabetes a CHD equivalent (104,114).
The evidence is unequivocal that an abnormal lipid profile is a significant risk factor for atherosclerotic macrovascular disease in patients with diabetes (115,116). Modification of lipid and other risk factors has clearly been shown to lower mortality due to cardiovascular disease (CVD) in the general population (117,118,119,120,121,122,123,124,125,126,127). This has underscored the need for management of CVD risk factors (in addition to control of hyperglycemia) in individuals with diabetes (114,128,129,130). The United Kingdom Prospective Diabetes Study (UKPDS) demonstrates that controlling lipids and blood pressure may be significantly more effective in preventing CVD than is controlling hyperglycemia (122,131). Thus, in individuals with diabetes, the management of lipid disorders, as well as total risk-factor management, is of paramount importance.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


