Hypersensitivity: Type I
Introduction
Hypersensitivity
The term hypersensivity is used to define a set of responses often categorized as types I–IV. These designations were first designated by Coombs and Gell in the early 1960s, as discussed below. In fact, the immune responses associated with each of the Coombs–Gell hypersensitivity designations manifest as a result of exaggerated normal responses that have been described in earlier chapters. Thus, the cellular and molecular mechanisms of such reactions are virtually identical to normal host defense responses. Unfortunately, under some circumstances, these immune responses produce damaging and sometimes fatal results, hence, hypersensitivity to the antigens stimulating these responses. They cause immune-mediated damage to the host because they manifest as exaggerated reactions to foreign antigens or inappropriate reactions to self-antigens.
Coombs–Gell Hypersensitivity Designations
In the early 1960s, hypersensitivity reactions were divided into four types, designated I–IV by Coombs and Gell; these are summarized below. Although the lines of distinction used to separate these four types of hypersensitivity have blurred through the years as our knowledge of cellular and molecular immunology has grown, the Coombs–Gell designations are still relevant.
Type I. IgE-mediated reactions (commonly called allergic reactions or allergy) are stimulated by the binding of IgE, via its Fc region, to high-affinity IgE-specific Fc receptors designated FcεRI. As we shall see later on in this chapter, FcεRI are expressed on mast cells, basophils, and eosinophils. Binding of antigen to preformed antigen-specific IgE bound to high-affinity FcεRI initiates type I hypersensitivity reactions. When the IgE molecules encounter antigens, a cascade of events leads to destabilization and release of inflammatory mediators and cytokines from mast cells and basophils. This ultimately results in the clinical manifestations of type I hypersensitivity, which include rhinitis, asthma, and, in severe cases, anaphylaxis (from the Greek ana, which means “away from,” and phylaxis, which means “protection”). Type I hypersensitivity reactions are rapid, occurring within minutes after challenge (reexposure to antigen). Consequently, allergic reactions are also called immediate hypersensitivity.
Type II. Type II hypersensitivity is the result of humoral cytolytic or cytotoxic reactions that occur when IgM or IgG antibodies bind inappropriately to antigen on the surface of cells and activate the complement cascade. This culminates in the destruction of the cells. Type II hypersensitivity is discussed in detail in Chapter 16.
Type III. Type III hypersensitivity, also known as immune complex reactions occur when antigen-IgM or antigen-IgG complexes accumulate in the circulation or in tissue and activate the complement cascade. Granulocytes are attracted to the site of activation, and damage results from the release of lytic enzymes from their granules. Reactions occur within hours of challenge with antigen. Type III hypersensitivity is also discussed in detail in Chapter 16.
Type IV. Cell-mediated reactions—commonly called delayed-type hypersensitivity (DTH)—are mediated by T-cell-dependent effector mechanisms involving both CD4+ TH1 cells and CD8+ cytotoxic T cells. Antibodies do not play a role in type IV hypersensitivity reactions. The activated TH1 cells release cytokines that cause accumulation and activation of macrophages, which in turn cause local damage. This type of reaction has a delayed onset that may occur days or weeks after challenge with antigen. Type IV hypersensitivity is discussed in detail in Chapter 17.
General Characteristics of Allergic Reactions
The sequence of events involved in the development of allergic reactions can be divided into several phases: (1) the sensitization phase, during which IgE antibody is produced in response to an antigenic stimulus and binds to specific receptors on mast cells and basophils; (2) the activation phase, during which reexposure (challenge) to antigen triggers the mast cells and basophils to respond by release of the contents of their granules; and (3) the effector phase, during which a complex response occurs as a result of the effects of the many inflammatory mediators released by the mast cells and basophils. As noted above, the clinical manifestations of these effector mechanisms include rhinitis, asthma, and anaphylaxis.
Sensitization Phase
The immunoglobulin responsible for allergic reactions is IgE. All normal individuals can make IgE antibody specific for a variety of antigens when antigen is introduced parenterally (entering the body via subcutaneous, intramuscular, or intravenous routes, but not through the alimentary tract) in the appropriate manner. However, as will be discussed below, some individuals are genetically predisposed to certain allergies. Note that allergic reactions can be elicited not only upon reexposure to the same antigen that initiated IgE synthesis, but also to other antigens that have the same epitopes. Sensitization to allergens can occur through any means, including skin contact, ingestion, injection, and inhalation. Approximately 50% of the population generates an IgE response to airborne antigens that are encountered only on mucosal surfaces, such as the lining of the nose and lungs and the conjunctiva of the eyes. However, after repeated exposure to a large number of airborne allergens such as plant pollens, mold spores, house dust mites, and animal dander, approximately 20% of the general population develops clinical symptoms, resulting in seasonal or perennial allergic rhinitis. An outdated yet commonly used term used to describe the clinical symptoms induced by airborne allergens is hay fever.
The term atopy (from the Greek word atopos meaning “out of place”) is frequently used to refer to IgE-mediated hypersensitivity and the adjective atopic to describe affected patients. Children of atopic individuals often suffer from allergies themselves, indicating that familial tendencies are common. Evidence suggests that IgE responses are genetically controlled by major histocompatibility complex (MHC)-linked genes located on chromosome 6. Recently, other IgE regulatory genes have been implicated, including the high-affinity IgE Fc receptor (FcεRI) gene on chromosome 11 and the TH2 IL-4 gene cluster on chromosome 5, which contains genes for IL-3, IL-4, IL-5, IL-9, and IL-13.
TH2 Cell Dependency of IgE Antibody Production
Several lines of evidence have demonstrated that IgE antibody production is TH2 cell dependent. The mechanism by which these cells promote B-cell isotype switching has not been fully elucidated, although it is clear that certain cytokines produced by TH2 cells, most notably IL-4 and IL-13, play a pivotal role. The administration of neutralizing antibodies to IL-4 in mice inhibits IgE production. In addition, IL-4 knockout mice cannot produce IgE following infection with Nippostrongylus brasiliensis, a nematode that induces high IgE responses in normal mice. A comparison of IL-4 levels in allergic versus nonallergic people has shown that IL-4 levels are significantly higher in the allergic population. Consistent with this observation, IgE levels are approximately tenfold higher in allergic individuals. It is important to note, however, that serum IgE in normal and allergic individuals represents only a fraction of total IgE in an individual since most of the IgE is bound to cells expressing high affinity FcεRI. In normal individuals, the concentration of serum IgE is the lowest of all immunoglobulins. It has been suggested that the low levels of IgE antibody in nonallergic individuals are maintained by suppressor effects mediated by IFN-γ produced by TH1 cells, which downregulates IgE production. Thus, in normal individuals, a balance is maintained between TH2-derived cytokines, which upregulate IgE responses, and TH1-derived cytokines, which downregulate IgE responses. Natural events such as infections with certain pathogens may disturb this balance and stimulate IgE-producing B cells. Therefore, allergic sensitization may result from failure of a control mechanism, leading to overproduction of IL-4 by TH2 cells and, ultimately, increased IgE production by B cells. Once adequate exposure to the allergen has been achieved by repeated mucosal contact, ingestion, or parenteral injection, resulting in the production of IgE antibody, an individual is considered to be sensitized. Once IgE antibody is made and secreted by allergen-stimulated B cells, it rapidly attaches to mast cells and basophils as it circulates past them.
Mast cells, the main effector cells responsible for allergic reactions, are a ubiquitous family of cells generally found around blood vessels in the connective tissue, in the lining of the gut, and in the lungs. They are large mononuclear cells that are heavily granulated and deeply stained by basic dyes (see Figure 15.1). Mast cells are derived from progenitor cells that migrate to tissues (e.g., connective tissue and epithelium), where they differentiate into mature mast cells. In some species, including humans, circulating basophils also take part in allergic responses and function in much the same way as tissue-based mast cells. Unlike mast cells, basophils mature in the bone marrow and are present in the circulation in their differentiated form. One of the most important features that mast cells and basophils share is receptors (FcεRI) on their cell membranes that bind with high affinity to the Fc portion of IgE. Once bound, the IgE molecules persist at the cell surface for weeks. The cell will remain sensitized as long as enough IgE antibody remains attached; the IgE molecules will trigger the activation of the cell when it comes into contact with antigen. The mast cell is not specific for any particular antigen; the IgE bound to it is.
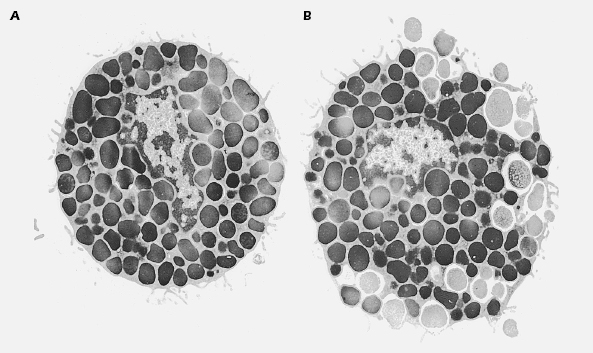
Sensitization may also be achieved passively, by transfer of serum that contains IgE antibody to a specific antigen. A procedure of historical interest only, known as the Prausnitz-Kustner (P-K) test, was performed as a test for the antibodies responsible for anaphylactic reactions. Serum from an allergic individual was injected into the skin of a nonallergic person. After 1–2 days, during which the locally injected antibody diffused toward neighboring mast cells and became bound to them, the site of injection was said to be sensitized, and would respond with an urticarial reaction (hives) when injected with the antigen to which the donor was allergic. Such a reaction in passively sensitized animals is called passive cutaneous anaphylaxis (PCA).
Activation Phase
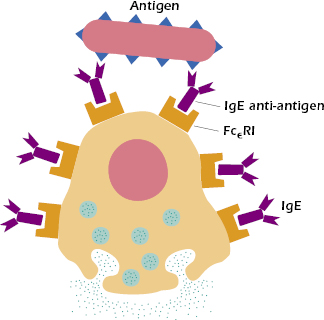
The activation phase of allergic reactions begins when the mast cell is triggered to release its granules and their inflammatory mediators. At least two of the FcεRIs must be bridged together in a stable configuration for the activation phase to occur. In the simplest and most immunologically relevant manner, this linkage is accomplished by a multivalent antigen that can bind a different molecule of IgE to each of several epitopes, thus cross-linking them on the mast cell surface and effectively triggering the cell to degranulate (see Figure 15.2). The physiologic consequences of IgE-mediated mast-cell degranulation depend on the dose of antigen and route of entry. Mast cells that degranulate within the gastrointestinal tract cause increased fluid secretion and peristalsis, which, in turn, can result in diarrhea and vomiting. In contrast, degranulation of mast cells in the lung causes a decrease in airway diameters and increased mucus secretion. These events lead to congestion and blockage of the airways (coughing, wheezing, phlegm), and to swelling and mucus secretion in nasal passages. Finally, degranulation of mast cells present along the blood vessels causes increased blood flow and vascular permeability, resulting in increased fluid in tissues or edema. This causes increased flow of lymph from the local lymph nodes, which in turn leads to increased numbers of cells and increased protein in tissue, all of which contribute to the inflammatory response.
Cross-linking of FcεRI receptors may also be accomplished in other experimentally useful ways, such as the addition of an antibody specific for IgE or FcεRI. IgE receptor molecules on the surface of mast cells (Figure 15.3A), exposure to sugar-binding lectins (Figure 15.3B), or the use of chemical cross-linkers (Figure 15.3C) or antibodies against FcεRI (Figure 15.3D). As expected, dimers or aggregates of IgE will also cross-link these Fc receptors and activate mast cells to degranulate. Finally, activation of mast cells can also be achieved using calcium ionophores, which induce a rapid influx of calcium ions into the cell, triggering the signaling cascade leading to degranulation.
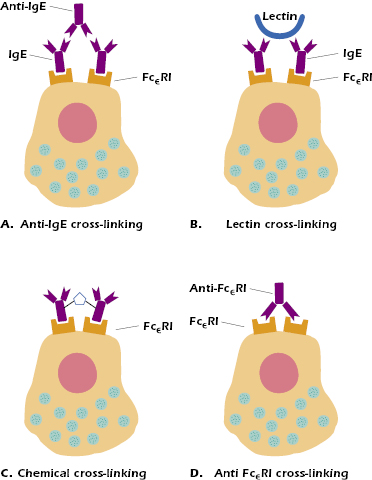
Mast cells may also be activated naturally through mechanisms other than IgE Fc receptor cross-linking. Anaphylactoid reactions are produced by the anaphylatoxins C3a and C5a (products of complement activation; see Chapter 14), as well as by various drugs such as codeine, morphine, and iodinated radiocontrast dyes. Physical factors such as heat, cold, or pressure can also activate mast cells; for example, cold-induced urticaria is an anaphylactic rash induced in certain individuals by chilling an area of skin. Another example is dermatographic uticaria in which the skin becomes raised and inflamed when stroked, scratched, or slapped. Finally, as noted above, certain lectins (sugar-binding molecules) can also cross-link IgE Fc receptors (see Figure 15.3B). High concentrations of lectins are found in certain foods, such as strawberries. This might explain the urticaria induced in some individuals after eating these foods.
The triggering of a mast cell by the bridging of its receptors initiates a rapid and complex series of events culminating in the degranulation of the mast cell and the release of potent inflammatory mediators. Because of the ease with which its outcome can be measured, the mast cell has served as a model for the study of activation of cells in general. Among the events known to occur rapidly are receptor aggregation and changes in membrane fluidity; these result from methylation of phospholipids, which leads to a transient increase in intracellular levels of cyclic adenosine monophosphate (cAMP) followed by an influx of Ca2+ ions. The intracellular levels of cAMP and cyclic guanosine monophosphate (cGMP) are important in the regulation of subsequent events. In general, a sustained increase in intracellular cAMP at this stage will slow, or even stop, the process of degranulation. Thus, activation of adenylate cyclase, the enzyme that converts adenosine triphosphate (ATP) to cAMP, provides an important mechanism for controlling anaphylactic events.
As noted earlier, allergic reactions are often referred to as immediate hypersensitivity. This term is appropriate in light of the very rapid consequences of IgE Fc receptor cross-linking, beginning with microfilament transport of mast-cell granules to the cell surface. Once at the cell surface, granule membranes fuse with the cell membrane and the contents are released to the exterior via exocytosis (see Figure 15.1B). Depending on the extent of cross-linking on the cell surface, any cell can release some or all of its granules. This explosive release of granules is physiologic and does not imply lysis or death of the cell. In fact, the degranulated cells regenerate; once the contents of the granules have been synthesized, the cells are ready to resume their function.
Effector Phase
The symptoms of allergic reactions are entirely attributable to the inflammatory mediators released by the activated mast cells. It is helpful to place these mediators in two major categories (Figure 15.4). The first category consists of basic preformed mediators, which are stored in the granules by electrostatic attraction to a matrix protein and are released as a result of the influx of ions, primarily Na+ (Figure 15.4A). Cytokines released from mast cells undergoing degranulation, including IL-3, IL-4, IL-5, IL-8, IL-9, tumor necrosis factor (TNF)-α, and GM-CSF (granulocyte-macrophage–colony stimulating factor), also play a role in attracting and activating inflammatory cells locally. Inflammatory cells participate in the so-called late-phase reactions of allergic responses (described later in this chapter) in concert with the second category of mast cell mediators—those synthesized de novo. The second category, newly formed mast cell mediators, consists of substances synthesized, in part, from membrane lipids (Figure 15.4B). Many potent substances are released during degranulation; only the most important members of each category are considered here.
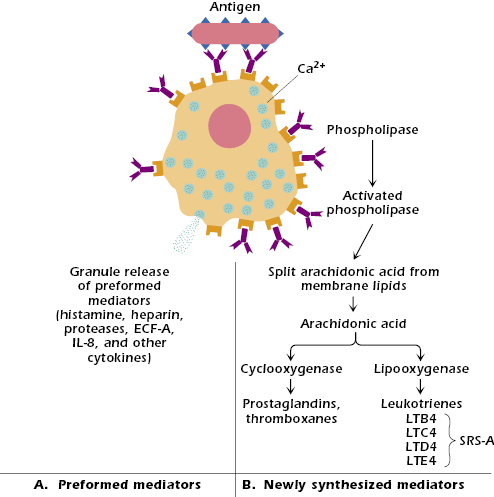
Preformed Mediators
Histamine. Histamine is formed in the cell by decarboxylation of the amino acid histidine; it is stored in the cell by binding via electrostatic interaction to an acid matrix protein called heparin. When released, histamine binds rapidly to a variety of cells via two major types of receptor, H1 and H2, which have different tissue distribution and mediate different effects. When histamine binds to H1 receptors in smooth muscles, it causes constriction; when it binds to H1 receptors on endothelial cells, it causes separation at their junctions, resulting in vascular permeability. H2 receptors are involved in mucus secretion, increased vascular permeability, and the release of acid from stomach mucosa. All these effects are responsible for some of the major signs of systemic anaphylaxis: difficulty in breathing (asthma) or asphyxiation result from the constriction of smooth muscle around the bronchi in the lung, and the drop in blood pressure is a consequence of the extravasation of fluid into tissue spaces as the permeability of blood vessels increases. H1 receptors are blocked by antihistamines, such as Benadryl®
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


