Resistance and Immunization to Infectious Diseases
Introduction
By now, the reader should fully appreciate that the primary function of the immune system is to defend the body against diseases caused by pathogens. Historically, infectious diseases have been the leading cause of death for human populations, with most deaths occurring in infancy and childhood. There have been many catastrophic epidemics of infectious diseases in history. For example, the bubonic plague caused by the bacterium Yersinia pestis killed one quarter of the European population in the mid-1300s. Hence infectious diseases have provided tremendous selective pressures for the evolution of the immune system. As we have discussed in the early chapters of this book, host defenses are characterized by a considerable amount of layering and redundancy. Layering refers to defense in depth and includes physical barriers, such as the skin and mucosal membranes, and innate and adaptive immune mechanisms. Redundancy is exemplified by the fact that there are several types of phagocytic cells, antigen-presenting cells (APCs), cytokine-producing cells, opsonins, and so on, such that for many immune functions, multiple mechanisms are in place to achieve the desired end. The redundancy of the immune system allows the host to survive for prolonged times, despite severe immune impairment. For example, some individuals with deficiencies in some immunoglobulin isotypes (e.g., IgA deficiency, the most common Ig deficiency) are thought to live a normal existence because other immunoglobulin classes compensate for the immune deficit.
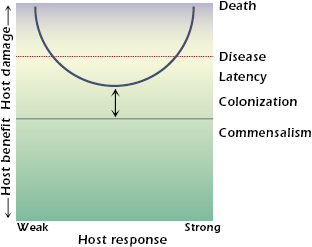
Microbes differ in their pathogenicity and virulence. Only a small minority of all microorganisms on earth are pathogens for humans. Pathogens are defined as microbes capable of causing host damage. Host damage can occur at the cellular, tissue, or organ level. When host damage reaches a certain threshold, it can manifest itself as disease. If sufficient damage occurs, the death of the host can be the outcome. Host damage can result from a variety of mechanisms and can be mediated by the microbe, the host, or both. Examples of mechanisms of microbe-mediated damage include the production of toxins, cellular apoptosis resulting in depletion of immune cells, and the elaboration of enzymes that cause tissue necrosis. Examples of mechanisms of host-mediated damage include destructive inflammation, fibrosis, and autoimmunity. The realization that host damage is the relevant parameter by which to characterize the outcome of the host–pathogen interaction is the basis of the recently proposed damage-response framework of microbial pathogenesis. According to this conceptual framework, the term pathogenicity is defined as the capacity of a microbe to cause damage in a host, and virulence is defined by the relative capacity of a microbe to cause damage in a host. Virulence and pathogenicity are not singular microbial properties, because they can be expressed in only susceptible hosts and reflect complex interactions among hosts; microbes; and myriad environmental, social, and human factors. The damage-response framework is illustrated in Figure 21.1. The host response is represented by a continuum from weak to strong. Weak responses are those that are insufficient, poor, or inappropriate. In other words, they are not strong enough to benefit the host. Strong responses are those that are excessive, overly robust, or inappropriate, and therefore have the ability to cause damage to the host. When a threshold amount of damage is reached, the host will show symptoms of disease. If damage is severe, death can ensue.
Humans harbor many species of microbes. When a human host encounters a microbe, the interaction can result in one of two outcomes: elimination or infection. Elimination can occur when the host–microbe encounter does not result in the establishment of the microbe in the host. Infection is the acquisition of a microbe by a host. Note that although the term infection is often used synonymously with the word disease, the two are not the same. Infection is followed by one of five outcomes: (1) elimination, (2) commensalism, (3) colonization, (4) persistence (or latency), or (5) disease. In the latter four outcomes, the relationship between the host and microbe is maintained, but the amount of damage sustained by the host differs (Figure 21.2). Elimination can follow infection as a result of action by host defense mechanisms or therapeutic intervention. Neither commensalism nor colonization rarely, if ever, results in symptomatic or clinically evident host damage, but these states can differ in the amount of host damage and in their ability to progress to disease. When there is no host damage, commensalism and colonization are essentially indistinguishable states.
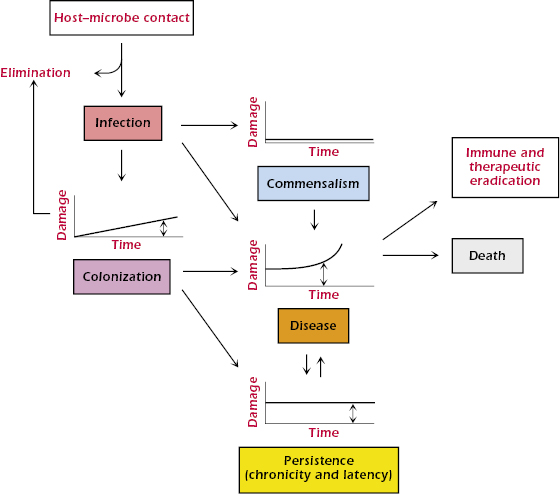
Colonization is a term that is usually used for microbes with significant pathogenic potential that establish themselves in the host without causing symptoms. Colonization can lead to elimination, persistence, or disease, depending on the competency of host defenses, the virulence of the microbe, and the effectiveness of the immune response. Many colonizing events stimulate immune responses and prevent future infection and/or disease caused by the relevant microbe. For example, a microbe with high pathogenic potential may establish itself on a mucous membrane, cause a small amount of damage that is insufficient to cause clinical symptoms, and elicit an immune response that eradicates it. In that situation, colonization immunized the host against reacquisition of the microbe.
Persistence (or latency) is a state whereby microbes take up residence in the host and cannot be eradicated, despite causing host damage. For example, in many humans, Mycobacterium tuberculosis infection is not symptomatic, even when the microbe has established itself in the host and is able to survive for long periods of time in a granuloma (see Chapter 17). In this state of persistence, local tissue damage and alterations in normal tissue occur due to granuloma formation, but the damage is not sufficient to produce clinical disease. However, unlike colonization, the host defense mechanisms cannot eradicate the mycobacteria, and the infection becomes persistent. In most individuals, this state is maintained with the infection confined to a granuloma. However, in some individuals, this state progresses to tuberculosis, the disease caused by M. tuberculosis. In the damage-response framework, disease occurs when the presence of the microbe in the host results in sufficient damage to manifest clinical symptoms.
In individuals with intact immune systems, microbes must be sufficiently virulent to establish themselves and cause infection. However, in individuals with compromised immune systems, low-virulence microbes can cause serious infections. Microbes that are pathogenic only in individuals with weakened immune systems are often referred to as opportunistic pathogens. Hence the microbial properties of pathogenicity and virulence are associated with, and partially depend upon, the immune status of the host. In circumstances of normal immune function, commensal microorganisms are not harmful, and some serve important roles for the host, such as the production of vitamin K by gut bacteria. However, commensals can become pathogens in circumstances in which the normal host defenses are breached. For example, both Staphylococcus epidermitis and Candida albicans are part of the normal skin flora but can cause life-threatening infections in patients with intravenous catheters that provide inappropriate access of these organisms to the circulatory system because of a break in the skin. Some therapies for cancer produce immune suppression, which leaves patients at risk for serious infections with low-virulence microbes, such as commensals.
In this chapter, we will discuss how mammalian hosts protect themselves against various types of pathogens and how the immune system can be primed for antimicrobial defense through active and passive immunization. It is assumed that the reader is now familiar with the principal concepts underlying innate and adaptive immune defenses.
Host Defense Against the Various Classes of Microbial Pathogens
The most effective immune response to a particular microbe varies with the type of pathogen and the microbial strategy for pathogenesis. Viruses, bacteria, parasites, and fungi each use different strategies to establish themselves in the host, and, consequently, the effective immune response for each of these classes of microbial pathogens is different. Although each pathogen is different, certain themes emerge when one considers immune responses to the various classes of pathogens.
Immunity to Viruses
All viruses are obligate intracellular pathogens, and many have evolved to have highly sophisticated mechanisms for cellular invasion, replication, and evasion of the immune system. Host defenses against viral infections aim to first slow viral replication and then to eradicate infection. The antiviral response can be complex, with several factors affecting the outcome of the host–pathogen interaction, such as the route of entry, site of attachment, aspects of pathogenesis by the infecting virus, induction of cytokines, antibody response, and cell-mediated immunity. An important early defense mechanism consists of the production of various types of interferons (IFNs) including IFN-α by leukocytes, IFN-β by fibroblasts, and INF-γ by T and natural killer (NK) cells. Interferons are antiviral proteins, or glycoproteins, produced by several different types of cell in the mammalian host in response to viral infection (or other inducers, such as double-stranded RNA). Interferons serve as an early protective mechanism. IFN-α and IFN-β produced by virally infected cells diffuse to adjoining cells and activate genes that interfere with viral replication. These interferons also stimulate production of MHC class I molecules and proteasome proteins that enhance the ability of virally infected cells to present viral peptides to T cells. Furthermore, IFN-α and IFN-β activate NK cells that recognize and kill host cells infected with viruses, thus limiting viral production.
NK cells are characterized by their ability to kill certain tumor cells in vitro without prior sensitization and constitute an early cellular defense against viruses. Later in the course of infection, when antibodies to viral antigens are available, NK cells can eliminate host cells infected with virus through antibody-dependent cell-mediated cytotoxicity (ADCC). NK cells also produce INF-γ, a potent activator of macrophage function that helps prime the immune system for producing an adaptive immune response. Complement system proteins damage the envelope of some viruses, which may provide some measure of protection against certain viral infections.
While the innate immune mechanisms that include IFNs, NK cells, and possibly complement, function to slow and partially contain many viral infections, the infection may progress, with viral replication and damage triggering an adaptive immune response. The humoral response results in the production of antibodies to viral proteins. Some antibodies can prevent viruses from invading other cells, and these are called neutralizing antibodies. IgG appears to be the most active isotype against viruses. Opsonization represents a convergence of humoral and cellular immune mechanisms. IgG, which has combined with viral antigens on the surface of infected host cells through its Fab region, also links to Fc receptors on several cell populations, including NK cells, macrophages, and polymorphonuclear (PMN) cells. These cells then can phagocytose and/or damage the virus-infected cell through ADCC.
Antibodies to viral proteins can prevent infection by interfering with the binding of virus to host cells. The production of secretory IgA can protect the host by preventing infection of epithelial cells in mucosal surfaces. Antibodies can also interfere with the progression of viral infection by agglutinating viral particles, activating complement on viral surfaces, and promoting phagocytosis of viral particles by macrophages. The production of an antibody response serves to limit viral spread and facilitates the destruction of infected host cells by ADCC. Hence the effective antibody response to viruses include the production of antibodies that
- Neutralize (or impede) the infectivity of viruses for susceptible host cells
- Fix complement and promote complement damage to virions
- Inhibit viral enzymes
- Promote opsonization of viral particles
- Promote ADCC of virus-infected cells
Different types of antibodies may be necessary for the control of specific types of viral infections. Consider the case of influenza and measles virus infections. Infection of the epithelium of the respiratory tract by influenza virus leads to production of virus in epithelial cells and spread of the virus to adjacent epithelial cells. An appropriate and sufficient immune response would involve the action of antibody at the epithelial surface. This action might be effected through locally secreted IgA or local extravasation of IgG or IgM. On the other hand, some viral diseases, such as measles and poliomyelitis, begin by infection at a mucosal epithelium (respiratory and intestinal, respectively) but exhibit their major pathogenic effects after being spread hematogenously to other target tissues. Antibody at the epithelial surface could protect against the virus, but circulating antibody could do likewise.
However, once a virus has attached to a host cell, it is usually not displaced by antibody. Hence an effective antibody response is usually not sufficient to eliminate a viral infection, particularly when the virus has established itself inside host cells. The eradication of an established viral infection usually requires an effective cell-mediated response. The adaptive cellular response results in the production of specific CD4+ and CD8+ T cells that are essential for clearance of viral infections. CD4+ T cells are believed to be intimately involved in the generation of effective antibody responses by facilitating isotype class switching and affinity maturation (see Chapter 11). CD4+ T cells also produce important cytokines to stimulate inflammatory responses at sites of viral infection and activate macrophage function. Cytotoxic lymphocytes CD8+ T cells (CTLs) are the principal effector T cells against viruses. They are generated early in viral infection and usually appear before neutralizing antibody. CD8+ T cells can recognize viral antigens in the context of major histocompatibility complex (MHC) class I molecules and can kill host cells harboring viruses. Since MHC class I molecules are expressed by most cell types in the host, CD8+ T cells can recognize many types of infected cells and thus represent a critically important component of the host adaptive response against viral infections. However, for certain noncytopathic viruses, such as hepatitis B, CD8+ T cells can be responsible for tissue injury. Hence, chronic hepatitis B infection results in persistent inflammation and damage to liver cells, resulting in fibrosis that can progress to organ failure.
In summary, innate immune mechanisms initially interfere with viral infection through the production of IFNs and the killing of infected cells by NK cells. These early defenses buy time until powerful, pathogen-specific adaptive immune responses are generated. The adaptive immune responses produce neutralizing antibodies that reduce the number of viral particles and CTLs that kill infected cells. The presence of neutralizing antibody would then protect against subsequent exposure to the same virus.
Immunity to Bacteria
Host protection against bacterial pathogens is achieved through a variety of mechanisms that include both humoral and cellular-mediated immunity. Antibacterial defenses include bacterial lysis, via antibody and complement, opsonization, and phagocytosis, with elimination of phagocytosed bacteria by the liver, spleen, and other components of the reticuloendothelial system. Bacteria and their products are internalized by APCs such as macrophages and dendritic cells, and processed for antigen presentation to T cells. Peptides resulting from such processing are presented to CD4+ T cells in the context of MHC class II molecules (see Chapter 10). This process amplifies the host response as T cells produce cytokines that activate macrophages and facilitate recruitment of additional inflammatory cells. The relative efficacy of the various immune mechanisms depends on the type of bacteria and, in particular, the cell-surface properties of the bacteria. Bacterial pathogens can be roughly divided into four classes: (1) Gram-positive, (2) Gram-negative, (3) mycobacteria, and (4) spirochetes, depending on their cell wall and membrane composition. Some Gram-positive and Gram-negative bacteria have polysaccharide capsules. Another crucial distinction between bacterial pathogens is whether they are intracellular or extracellular pathogens. Intracellular bacterial pathogens reside in cells and are partially shielded from the full array of host immune defenses, whereas extracellular bacterial pathogens are found outside cells. In general, humoral immunity is very important for protection against extracellular bacteria, whereas cellular immunity is the primary immune mechanism for the control and eradication of intracellular bacteria. TH17 cells are believed to play a crucial role in this host defense mechanism.
Gram-Positive Bacteria. Gram-positive bacteria have thick electron-dense cell walls composed of complex cross-linked peptidoglycan that allow them to retain the stain crystal violet (hence Gram-positive). In addition to a thick layer of peptidoglycan, the cell wall of Gram-positive bacteria contains teichoic acids, carbohydrates, and proteins. Teichoic acids are immunogenic and constitute major antigenic determinants of Gram-positive bacteria. This type of cell wall provides the Gram-positive bacteria with a thick layer of protection that makes them resistant to lysis by the complement system. Defenses against Gram-positive bacteria include specific antibody responses to provide opsonins and phagocytic cells, such as neutrophils and macrophages, to ingest and kill them. Opsonization and phagocytosis involve the action of IgG alone or in concert with C3b. The alternative complement pathway may be triggered directly by the Gram-positive bacterial cell wall, resulting in the deposition of complement opsonins in the cell surface and the production of mediators of the inflammatory response. Although the complement system does not lyse Gram-positive bacteria directly, it provides opsonins and mediators of inflammation that are critical for host defense.
Gram-Negative Bacteria. Gram-negative bacteria do not retain crystal violet stain and have a layered cell-wall structure composed of outer and inner membranes separated by a thin layer of peptidoglycan in the periplasmic space. Hence Gram-positive and Gram-negative bacteria have major differences in their cell-wall structure. The outer membrane of Gram-negative bacteria contains lipopolysaccharide (LPS), which is also known as endotoxin. The polysaccharide portion of LPS has antigenic determinants that confer antigenic specificity. Many Gram-negative bacterial species include variants with different LPS structures that can be identified serologically as serotypes. LPS is highly toxic to humans and can produce cardiovascular collapse, hypotension, and shock during infection with Gram-negative bacteria. The alternative complement pathway may be activated directly by the LPS found in the walls of Gram-negative bacteria or by the polysaccharide capsule of Gram-negative bacteria acting on C3. Activation of the alternative pathway leads to the generation of the chemotactic molecules C3a and C5a and the opsonin C3b and can result in bacteriolytic action by the C5C9 membrane attack complex (see Chapter 14). The ability of the complement system to lyse some Gram-negative bacteria directly is an important distinction from Gram-positive bacteria, which are impervious to complement-mediated lysis because of the thick peptidoglycan layer. Defenses against Gram-negative bacteria include the complement system, specific antibody, and phagocytic cells.
Mycobacteria. Mycobacteria have cell walls distinct from Gram-positive and Gram-negative bacteria. Mycobacterial cell walls are characterized by a high lipid content, which makes the bacteria difficult to stain. A useful diagnostic microbiological property of the mycobacterial cell wall is acid fastness, which allows the cell wall to retain certain dyes after being treated with acid. Mycobacteria grow slowly and have hydrophobic surfaces that make them clump. Mycobacterial cell wall components elicit strong immune responses during infection, including delayed-type hypersensitivity (DTH) reactions that form the basis for the tuberculin test (see Chapter 17). Hypersensitivity reactions to mycobacterial proteins may be involved in the pathogenesis of mycobacterial infections. Mycobacteria elicit strong antibody responses but the protective role of humoral immunity is uncertain. The primary defense mechanisms against mycobacteria are macrophages and cell-mediated immunity.
Spirochetes. Spirochetes are thin helical microorganisms and include the etiologic agents of syphilis (Treponema pallidum) and Lyme disease (Borrelia burgdorferi). Spirochetes lack cell walls such as those found in Gram-positive bacteria, Gram-negative bacteria, and mycobacteria. Instead they have a thin outer membrane that contains few proteins. Spirochetes are thin, fragile, and require special techniques for visualization in the microscope, such as dark-field microscopy and immunofluorescence. Important host defenses against spirochetes include complement, specific antibody, and cell-mediated immunity.
Immunity to Parasites
The parasites are a diverse group of complex pathogens that include the multicellular helminths and single-celled protozoa. Nearly half of the world’s population harbors helminth infections. Many parasites have a variety of tissue stages that may differ in cellular location and antigenic composition, thus providing a difficult problem for the immune system. The diversity of parasites is such that it is difficult to make generalizations about effective host mechanisms that protect against parasitic diseases. However, it is clear that both innate and adaptive defense mechanisms are critically important for protection against parasitic infections. So-called type-2 immune responses dominate the adaptive immune mechanisms that confer protection to helminths as well as other parasites. The term “type-2 responses” is derived from the fact that these responses are characterized by the induction of TH2 responses. TH2 cells secrete cytokines such as IL-4, IL-5, IL-9, and IL-13 and display distinct transcription factor profiles (GATA-3, STAT-5, and STAT-6). It is noteworthy that although type-2 immune responses have been explored largely in the context of helminth infections and allergies (see Chapter 15), they are also induced by venoms, vaccine adjuvants such as alum, and some bacterial and viral infections. How such diverse stimuli trigger prototypic type-2 responses and the nature of the cellular and molecular networks that orchestrate these responses are still unclear.
Protozoa.
Protozoa may exist in either a metabolically active form called a trophozoite or a dormant tissue form known as a cyst. The protozoal diseases include amebiasis, malaria, leishmaniasis, trypanosomiasis, and toxoplasmosis. Host defenses against protozoa include both innate and adaptive humoral and cellular mechanisms, but their relative importance may vary with the individual pathogen. Some protozoal parasites, such as trypanosomes, are able to activate the complement system through the alternative pathway.
Complement activation combined with phagocytosis by neutrophils and macrophages of the innate immune system provide an important line of defense against many parasitic pathogens. For some protozoal infections such as amebiasis, malaria, and trypanosomiasis, humoral immunity in the form of antibody has been shown to mediate protection against infection. Vaccines capable of inducing protective antibody responses to each of these parasitic pathogens have not yet been developed. For other protozoal infections such as leishmaniasis and toxoplasmosis, cellular immunity is more important; again, there are currently no vaccines available.
Helminths.
Unlike the pathogenic protozoan microorganisms, the multicellular worms called helminths are large macroscopic pathogens that can range from 1 cm to 10 m. Their large size poses particular problems for host defenses, and the control of helminth infections requires a complex interplay between tissue and immune responses. The helminths are notorious for causing chronic infections that can elicit intense immune responses to worm antigens. There is general agreement that components of the innate immune system such as eosinophils and mast cells are important effector cells against helminths, but many aspects of the host response to worms remain obscure. IgE specific for helminth antigens is believed to be important for host defense by priming eosinophils for ADCC (see Figure 15.8). Worm infections are often accompanied by an increase in blood eosinophils and serum IgE levels.
Immunity to Fungi
Fungal pathogens are eukaryotes that tend to cause serious infections primarily in individuals with impaired immunity. Fungi cause tissue damage by the elaboration of proteolytic enzymes, inducing inflammatory responses. The most common fungal pathogen is Candida albicans. This organism is usually a harmless commensal, but it can cause disease in situations in which the normal defense mechanisms are compromised, such as breaks in skin resulting from intravenous catheters or surgery. Another group at risk for serious C. albicans-related diseases comprises individuals who have transient depletion of neutrophils as a result of chemotherapy. The fact that most cases of serious Candida infection require a break in the skin or a depletion in neutrophils suggests that innate defense mechanisms are largely responsible for preventing systemic diseases. However, patients with advanced human immunodeficiency virus (HIV) infection suffer from mucosal candidiasis, highlighting the importance of cell-mediated immunity in protection against this organism at the mucosal surfaces.
Other fungi, such as Histoplasma capsulatum and Cryptococcus neoformans, are acquired from the environment by inhalation in regions where the organism is found in soils. C. neoformans has a polysaccharide capsule that is required for virulence. Prevalence studies of asymptomatic individuals in such regions have shown a high incidence of infection with a very low incidence of disease, as evidenced by antibody responses or positive skin testing. Hence, for these organisms, it is likely that initial acquisition of the microbe results in an adaptive immune response that controls the infection. Some fungi, such as H. capsulatum, survive inside macrophages and are intracellular pathogens. The TH17 lineage of CD4+ T cells is believed to play an important role in host defenses against intracellular fungal infections.
Fungi differ from bacteria in that they have a different type of cell wall, composed of cross-linked polysaccharides. Fungal cells are generally impervious to lysis by the complement system. The host response to fungal infections includes both humoral and cellular responses. The primary form of host defense against fungal pathogens is widely acknowledged to be cell-mediated immunity. The need for intact T-cell function in resistance to fungi is particularly evident in the predisposition of patients with acquired immunodeficiency syndrome (AIDS) to life-threatening infections with such fungi as H. capsulatum and C. neoformans. Historically, antibody-mediated immunity was not thought to be very important against fungi, but several protective monoclonal antibodies have been described in recent years against C. albicans and C. neoformans. Hence it is likely that both cellular and humoral immune mechanisms contribute to protection against fungi.
Mechanisms by Which Pathogens Evade the Immune Response
Despite the formidable defense mechanisms of the intact immune system, some microorganisms manage to establish themselves in the host and cause life-threatening infections. These pathogenic microbes have special adaptations that allow them to evade the immune system or overcome immune responses. Learning about the mechanisms by which microbial pathogens evade the host immune response is important because it can teach us about the efficacy and limitations of host defense mechanisms. Furthermore, a better understanding of the strategies used by microbes to survive immune attack can be used to design new therapies and vaccines to fight infection.
Encapsulated Bacteria
Polysaccharide capsules are important virulence factors for several human pathogens, including Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae, Neisseria meningitidis (meningococcus), and Cryptococcus neoformans. These capsules are antiphagocytic and thus protect the pathogen from ingestion and killing by host phagocytic cells. Some capsules also interfere with the action of the complement system. Polysaccharide molecules are often weakly immunogenic, and infection by encapsulated pathogens may not necessarily elicit high titer antibody responses. Infants and young children (1–2 years of age) are particularly vulnerable to life-threatening infections with encapsulated bacteria because their immature immune systems do not mount adequate antibody responses. This is attributable, in part, to their lack of marginal B cells in the spleen. Other individuals at high risk are those with inherited or acquired deficiencies in antibody production and those who lack normal spleen function. Because the bacteria are cleared by the reticuloendothelial system in the spleen and liver, these organs are critical for protection against encapsulated pathogens. Individuals with compromised reticuloendothelial function as a result of disease (e.g., sickle cell anemia) or surgical removal of the spleen are particularly vulnerable to encapsulated bacteria. The mechanism of antibody action against encapsulated pathogens involves opsonins for phagocytosis and killing by neutrophils and macrophages. Antibodies to capsular polysaccharide function by promoting phagocytosis directly through Fc receptors or indirectly by activating complement. The generation of C3b following complement activation also facilitates opsonization for phagocytosis, which is another example of the redundancy of host defenses.
Toxins
For some bacterial infections, the manifestations of disease are caused by virulence factors called toxins. Bacterial toxins are proteins that produce their physiologic effects at minute concentrations. Examples of toxin-producing bacteria are Corynebacterium diphtheriae, Vibrio cholerae, and Clostridium tetani, the causes of diphtheria, cholera, and tetanus, respectively. Diphtheria is a condition in which C. diphtheriae replication and toxin production in the nasopharynx results in the formation of a tenacious membrane in the throat that can asphyxiate the patient. Cholera is a diarrheal illness caused by V. cholerae, resulting from toxin-mediated alteration of water resorption in the cells of the intestinal mucosa. Tetanus is a condition in which toxin produced by C. tetani produces unchecked excitation of peripheral muscles, resulting in titanic spasms. The relationship of the toxin function to bacterial invasion and evasion of the immune response is variable and may differ for each pathogen. Some toxins, such as tetanus and botulinum toxin, do not appear to injure the immune system directly. Diphtheria toxin may promote bacterial infection by damaging the mucosa. Bacillus anthracis, the causative agent of anthrax, produces pathogenic toxins that cause apoptosis in macrophages. The principle mechanism for this immune evasion involves a toxin called lethal toxin (LT). LT inhibits a macrophage protein kinase that is required for the transcription of antiapoptotic genes following cell activation.
Most toxins are highly immunogenic and elicit strong humoral and cellular immune responses. Specific antibodies can bind to and neutralize bacterial toxins. Protection against toxins is predominantly associated with IgG, although IgA may also be important in neutralization of certain exotoxins (e.g., secreted toxins) such as cholera enterotoxin. Because the exotoxins bind firmly to their target tissue, they generally cannot be displaced by subsequent administration of antitoxin (passive immunization using toxin-specific antibody and discussed later in this chapter). Hence in toxin-mediated diseases (e.g., diphtheria) prompt administration of antitoxin is necessary to prevent attachment of (additional) exotoxin and the damage caused by the toxin. This can be illustrated by the effectiveness of antitoxin given at varying times as protection against the lethal effects of diphtheria toxin in humans (Table 21.1). As the infection progresses, the effectiveness of administration of diphtheria antitoxin is significantly reduced. Some bacterial toxins are enzymes, such as the lecithinase of the bacterium Clostridium perfringens and snake venom. However, antibodies that bind the toxin may not necessarily inhibit the enzymatically active sites of toxin.
Table 21.1. Protection of Humans by Diphtheria Antitoxin Given on Indicated Day of Disease
| Day | Number of Cases | Fatality Rate |
|---|---|---|
| 1 | 225 | 9.0 |
| 2 | 1,445 | 4.2 |
| 3 | 1,600 | 11.1 |
| 4 | 1,276 | 17.3 |
| 5 (or later) | 1,645 | 18.7 |
Superantigens
The interaction of certain toxins with the immune system can have major immunologic consequences when they are able to bind the T-cell receptor (TCR) of large numbers of T cells. These toxins are known as superantigens and include the staphylococcal toxic shock syndrome. In the early 1980s, many cases of staphylococcal toxic shock syndrome were associated with tampon use in menstruating women. Since then, the frequency of the disease has decreased significantly in response to changes to the manufacture of tampons. Superantigens stimulate large numbers of T cells to proliferate, synthesize cytokines, and then die by apoptosis, resulting in the loss of important immune cells. The release of massive amounts of cytokines by the T cells (cytokine storm) is associated with hypotension, hypolemia, and organ failure, which can lead to death. B-cell superantigens that bind to and alter the expression of certain immunoglobulin gene families have also been described.
Antigenic Variation
Pathogens can escape the immune system by generating variants with different antigenic composition. This mechanism for evasion of host defenses is known as antigenic variation. Classical examples of pathogens that evade the host response by antigenic variation are influenza virus, HIV, Streptococcus pneumonia, trypanosomes, and Group A streptococcus. Each of these pathogens provides an illustrative example of a mechanism for antigenic variation. In the case of Group A streptococcus, the M protein is required for virulence and functions by preventing phagocytosis through a mechanism that involves deposition of fibrinogen on the bacterial surface. M proteins elicit protective antibody but are antigenically variable, so that streptococcal infection with one strain does not elicit resistance to other strains.
Influenza virus generates antigenic variation because it has a segmented RNA genome that can be resorted to yield virions expressing new combinations of the two main surface antigens: the hemagglutinin and neuraminidase surface proteins. Antigenic variation for influenza virus occurs through both antigenic drift and antigenic shift. Antigenic drift is the result of point mutations in the influenza virus genome; these mutations produce antigenic changes in the hemagglutinin and neuraminidase. Antigenic shift occurs when influenza virus expresses a new allele of hemagglutinin or neuraminidase protein that results in a major antigenic change and the emergence of a new viral strain. The result of antigenic drift and shift for the influenza virus is that the virus changes rapidly, and one influenza infection does not confer protection against subsequent infection. Furthermore, since each epidemic is antigenically different, a new vaccine against influenza must be reformulated every year.
HIV undergoes rapid antigenic variation in vivo because it has an error-prone reverse transcriptase that produces mutations that translates into antigenic changes in surface proteins. The problem of antigenic variation in HIV has been a major hurdle preventing the development of an effective vaccine. Other pathogens, such as Streptococcus pneumoniae
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


