HUMAN PRION DISEASES
SERGGIO C. LANATA, SVEN FORNER, AND MICHAEL D. GESCHWIND
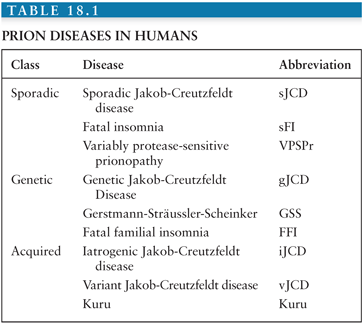
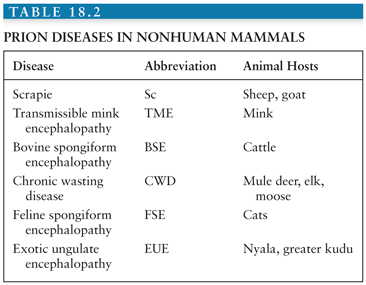
The transmissible spongiform encephalopathies (TSEs) are a group of closely related fatal neurodegenerative diseases that affect humans and several other mammals (Tables 18.1 and 18.2). TSEs have a few characteristics in common: (1) they are transmissible in nature and/or experimentally; (2) they invariably lead to brain dysfunction or “encephalopathy,” which manifests clinically as cognitive, behavioral, sensory, and/or motor dysfunction; (3) they are uniformly fatal; and (4) they currently can only be diagnosed definitively by neuropathology (or genetically in certain cases).
TSEs are caused by prions (for “proteinaceous infectious particles”), misshapen forms of the endogenous prion protein, in the central nervous system (1); hence, TSEs are now referred to as “prion diseases,” which is the term that will be used for this chapter. Prion (pree-ahn) diseases are unique in medicine in that they occur in three forms: sporadic, infectious/acquired, and genetic. All three mechanisms occur in humans. Prions are considered infectious because they rarely can be transmitted from person to person (iatrogenically) or even less commonly by ingestion. When they are transmitted into, or develop in, the nervous system, they cause the conversion of normal-shaped prion proteins into an abnormal, pathogenic conformation. This conversion process becomes exponential and leads to the spread of prions throughout the brain, causing neuronal dysfunction and cell death and, ultimately, neurodegeneration. Prions are intrinsically different from all other naturally occurring infectious agents, however, in that they do not rely on nucleic acid to replicate within the host organism. Furthermore, the presence of sporadic and heritable forms of prion disease also differentiates prions from all other infectious agents. This chapter will focus primarily on human prion diseases, with the caveat that most of our knowledge of the pathophysiologic mechanisms of prion disease has been obtained from animal and cell culture studies.
SCRAPIE, KURU, AND THE DISCOVERY OF PRIONS
The history of the discovery of prions is an inspiring journey of multidisciplinary scientific collaboration that profoundly changed our understanding of infectious and neurodegenerative diseases. This journey began with the careful study of the prion disease called scrapie, which primarily affects sheep and goats.
Scrapie is probably the earliest described prion disease, with the first reports published in 1750, but the disease was probably recognized as early as 1732 in Great Britain. The clinical signs of scrapie are variable; the most frequently observed clinical signs are emaciation, repetitive head rubbing, hyperesthesia, and pruritus. Pruritus is often severe enough to make the affected animal rub and scrape (hence the term “scrapie”) its body against rough surfaces, leading to wool loss and skin abrasions (2). Affected animals often become hypersensitive to outside stimuli, including noises, sudden movements, or handling by humans, to the extent some stimuli will cause the animal to startle violently or convulse. Other common signs include tremor and ataxia leading to impaired gait and falls. Behavioral changes are prominent and common. A nibbling response of the mouth is often evoked by scratching the animal’s back (“positive scratch response”). Scrapie has a long incubation period, ranging between 2 and 5 years, and affected animals live up to 6 months after symptom onset.
Scrapie is transmissible both naturally and experimentally. The natural means of transmission is a matter of debate, with both environmental and genetic susceptibility factors influencing transmission (3). Experimental transmissibility of scrapie, however, was firmly demonstrated in 1936 when Cuillé and Chelle (4) showed that healthy sheep inoculated with scrapie-infected spinal cord tissue developed the disease. At the time, however, the infectious agent of scrapie was unknown.
Puzzled by scrapie’s long incubation period and devastating course, Bjorn Sigurdsson, an Icelandic veterinarian, first proposed the notion of “slow viral infections” in the 1950s regarding a paralytic encephalitis of sheep called rida (5). In his view, these neurologic diseases were caused by unknown viruses with prolonged incubation periods (years to decades) that led to death after a short and progressive clinical course. Sigurdsson’s ideas shed light on the etiology of many diseases that were poorly understood at the time (6). For example, maedi, a deadly form of pneumonitis in sheep, and visna, a slowly progressive encephalitis, were found to be caused by the same virus, now known as the maedi-visna lentivirus (7). The suspected viral cause of scrapie, on the other hand, still remained elusive.
Concurrently, in the 1950s, D. Carleton Gajdusek, an American physician and virologist, initiated the scientific study of a human neurologic disease known as kuru in Papua New Guinea. Papua New Guinea is one of the most culturally diverse countries in the world, with more than 800 languages spoken among roughly 6.3 million people segregated in different tribes. The kuru epidemic emerged in the early twentieth century among the Fore tribe that inhabits one of the highland provinces. Kuru means to shiver from fever or cold in the language of the Fore people (8). The disease is characterized clinically by the insidious onset of progressive cerebellar ataxia, eventually leading to inability to walk. A typical early feature of the disease is the presence of unusual laughter, giving rise to the phrase “the laughing death” to describe kuru (9). In later stages, brainstem findings typically manifesting as eye movement abnormalities and severe dysarthria are also common. Like scrapie, the incubation period for kuru is long, as long as 50 years (10), and disease duration is relatively short, usually less than 12 months.
Early anthropologic studies of the Fore people described a ritualistic form of cannibalism that prevailed across all age groups. Fore people consumed the bodies of their deceased relatives (endocannibalism), including those who died of kuru (11). This was done with the hope of maintaining the spirit of the deceased within the village community. Perhaps partly for this reason, Gajdusek and his team initially suspected an infectious, probably viral, etiology to kuru, borrowing from Sigurdsson’s concept of slow viral infections (12). It soon became clear, however, that kuru was not a typical infectious disease, in that victims did not exhibit fever or other clinical signs of infection, including no signs of meningoencephalitis, no obvious cerebrospinal fluid (CSF) abnormalities, and patients did not respond to antibiotic medications. Furthermore, autopsy studies of brains affected by kuru did not demonstrate the inflammatory changes typically seen in many forms of nervous system infections. Additionally, it was not transmissible to small laboratory animals and no infectious agent had been identified (8). This led Gajdusek and others to conduct an exhaustive search of environmental factors that could explain the transmissible and seemingly infectious nature of kuru (13,14), but none were found. The absence of an environmental cause for kuru coupled with the well-described tribal and familial distribution of the disease led some to believe that kuru was a genetically inherited condition (15). Large epidemiologic studies showed that the incidence of kuru dramatically decreased among members of tribes who abolished endocannibalism, thus pointing toward a transmissible disease mechanism rather than a genetic one (8).
A major breakthrough came when William J. Hadlow, a veterinarian pathologist, made note of the neuropathologic similarities between kuru and scrapie and postulated that a similar infectious agent was the cause of both diseases (16). This was an important conceptual contribution to the study of prion diseases in general, as prior to the 1950s, the efforts to understand scrapie and kuru were disjoined (1). This observation further motivated scientists to take on the challenge of identifying the causative agent of these diseases.
Toward this goal, a series of careful experiments done on scrapie-infected brains demonstrated that the scrapie agent was resistant to conditions that normally inactivated other infectious organisms like viruses and bacteria, such as exposure to formalin (17), very high temperatures (18), and other solvents (19). Scrapie, and presumably kuru, was beginning to show properties that distinguished it from other infectious diseases.
Siggurdson’s and Hadlow’s hypotheses were eventually supported experimentally in 1966, when the transmission of kuru was demonstrated by Gajdusek and colleagues (20) through the intracerebral inoculation of diseased brain tissue in the chimpanzee. The proven transmissibility of kuru helped solidify the natural transmissibility theory that had already been gaining momentum based on anthropologic and epidemiologic observations: kuru was transmitted through endocannibalism (21). Furthermore, neuropathologic similarities between kuru and Jakob-Creutzfeldt disease (JCD) had been clearly recognized in the late 1950s (22). And the same group of investigators that demonstrated transmission of kuru to chimpanzees also demonstrated transmission of JCD to the chimpanzee in 1968 (23). This was an important experimental discovery, as JCD had previously been regarded as a human neurodegenerative disease of unknown etiology. Taken together, these observations led to the introduction of the term “transmissible spongiform encephalopathies” to highlight the many neuropathologic and clinical characteristics shared by these three diseases.
Still, although experimental transmissibility had been proven for scrapie, kuru, and JCD, and natural transmissibility had been shown for scrapie and kuru, the infectious substrate of these diseases remained unknown. As mentioned earlier, the scrapie agent did not share basic biochemical and biophysical properties with other known infectious organisms. This notion was strengthened in 1966, when Alper and colleagues (24) showed that extracts of scrapie-infected brains retained their infective nature even after exposure to large doses of ultraviolet light that were known to irreversibly damage the DNA and RNA molecules, thus implying that the scrapie agent lacked nucleic acid. Later, similar studies were conducted on tissue containing the agent responsible for kuru and JCD (25).
The idea of an infectious agent lacking nucleic acid was considered outlandish at the time. It implied that the agent that transmitted these diseases was, at its core, unlike any other known infectious agent. This idea captured the curiosity of neurologist Stanley Prusiner and other researchers. Through a series of careful experiments using animal models, Prusiner confirmed that the scrapie agent indeed resists inactivation by procedures that hydrolyze, modify, or shear nucleic acids; furthermore, Prusiner and colleagues demonstrated that the scrapie agent was inactivated by treatments that denatured protein (26). Based largely on this line of evidence, Prusiner postulated that a protein, which he named “prion” (pree-ahn) for “proteinaceous infectious particle,” was the principal carrier of scrapie (27).
The discovery of prions (PrP, for “prion protein”) shattered the concept that nucleic acids are the sole carriers of transmissible infectious disease. Alternative theories to explain the nature of the scrapie agent were proposed for years after Prusiner’s postulate (28,29), but none of these theories has been proven experimentally and the prion model has become widely accepted over the last two decades, supported by a vast body of evidence.
NORMAL AND PATHOGENIC PRION PROTEINS: PrPC AND PrPSc
After PrP was found to be the disease-producing substrate in concentrates of scrapie-infected brain tissue (27), the question arose whether PrP was acquired exclusively from the environment via an infectious mechanism or if it was also produced endogenously by the diseased animal.
In 1986, for the first time, researchers were able to isolate PrP-related genomic clones in normal hamsters using complementary DNA (cDNA) obtained from PrP messenger RNA (mRNA), thus demonstrating that mammalian cells carry the gene responsible for encoding PrP (30). Similar experiments were performed in mice (31) and other mammals including humans (32). The gene that encodes PrP is known as PRNP. PRNP encodes a normal form of PrP with incompletely understood cellular functions. The misshapen, pathogenic form of PrP, on the other hand, has unique biochemical properties that distinguish it from its normal counterpart (as will be discussed in the following sections). This distinction led to the current designation of “cellular PrP” (PrPC) to describe PrP in its normal form and “scrapie PrP” (PrPSc) to describe the misfolded, pathogenic form of PrP. The terms PrPC and PrPSc will be used accordingly in this chapter.
The Prion Protein Gene (PRNP)
The prion gene, PRNP, is located in the short arm of chromosome 20 in humans. It is composed of two exons and one intron, and the protein-coding region is located in exon 2. PRNP encodes the 253 amino acid PrP. A schematic of PRNP and the prion gene in other species is shown in Figure 18.1. There are more than 30 autosomal dominant mutations of PRNP that lead to inherited forms of prion disease in humans. These occur primarily in the form of point mutations as well as some insertion, and possibly deletion mutations. Point mutations normally occur in the central and C-terminal portions of PrP. Insertion mutations occur in the N-terminal end of the protein, which is normally composed of a nonapeptide followed by four octapeptide repeats. Different mutations lead to varied familial forms of the disease (33,34).
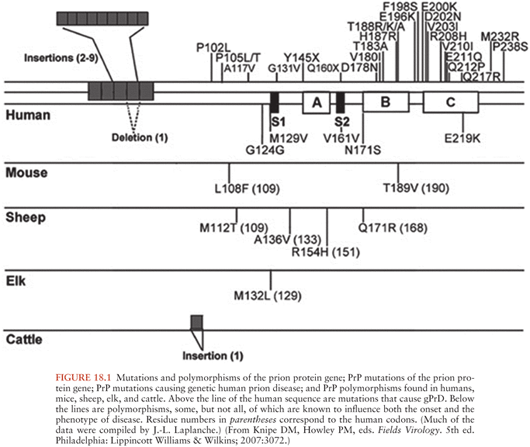
Historically, clinically-based studies of the heritable forms of prion disease in humans led to the identification of three classic phenotypes: Gerstmann-Sträussler-Scheinker disease (GSS), familial Jakob-Creutzfeldt disease (fJCD), and fatal familial insomnia (FFI). These three forms of familial JCD were described based on clinicopathologic features, prior to the identification of PRNP. The classification of familial (genetic) prion diseases, however, has changed with the division of familial forms based on specific PRNP mutations. Genetic prion diseases (gPrDs) are discussed in detail in the section, “Human Genetic Prion Diseases.”
Polymorphisms in the PRNP gene have been shown to influence prion disease susceptibility and the clinical phenotype. The most important polymorphism in humans is at amino acid 129 of PRNP, which can be methionine (M) or valine (V). As every person has two copies of each gene, humans can be either MM, MV, or VV. Homozygosity (either MM or VV) at codon 129 increases susceptibility to sJCD (35,36), for example. Likewise, in patients with gPrD, the cis codon 129 polymorphism (i.e., the codon 129 allele on the same DNA strand as the mutation) can greatly influence how a disease presents (37). The phenotypic influence of these polymorphisms will be discussed further under the gPrDs section.
The Function of Cellular PrP
PrPC is expressed mostly in the brain, but it has also been detected in a wide variety of mammalian tissues, including leukocytes, heart, skeletal muscle, lung, intestinal tract, spleen, and reproductive organs (38). The role of PrPC in the nervous system remains elusive and controversial. PrPC is a cell-surface glycoprotein that forms part of cell membrane structures known as lipid rafts (39). In the nervous system, lipid rafts are heavily involved in cellular signal transduction pathways, including neurotrophic factor signaling, cell adhesion and migration, axon guidance, myelin genesis, and synaptic transmission (40). It is therefore not surprising that most of the cellular functions of PrPC that have been described over the past two decades relate to a wide range of signal transduction pathways (41) involving molecular interactions with multiple cellular proteins (42), some of which are vital for normal neuronal function. For example, in mouse neuroblast brain cells, PrPC overexpression enhances cell proliferation and cell cycle reentrance, whereas PrPC silencing slows down the cell cycle; these effects are mediated via PrPC interactions with epidermal growth factor receptor (EGFR) in the plasma membrane (43). Similarly, PrPC expressed in pluripotent human embryonic stem cells have been shown to induce and modulate cell cycle dynamics and partly determine cell differentiation (44). PrPC has also been shown to be essential for myelin maintenance, as depletion of PrPC in neurons triggers a form of chronic demyelinating neuropathy in mice (45). PrPC has been implicated in neuroprotective and neurotoxic signaling cascades (46). Furthermore, there is an expanding body of evidence that supports clear interactions between PrPC and metal ions in different signaling pathways, particularly copper, zinc, and iron (47).
Despite the role of PrPC in a variety of important cellular functions (48), however, some PrPC knockout mice have been shown to develop normally and do not have signs of neurologic disease (49), thereby suggesting that the absence of PrPC is not a sufficient cause of neurodegeneration or neurodevelopmental abnormalities.
Structural Differences Between PrPC and PrPSc
All mammalian proteins are composed of a specific structure of amino acids (primary structure) that are arranged into identifiable repeating structures held together by hydrogen bonding (secondary structure), which in turn cause protein molecules to adopt specific three-dimensional conformations (tertiary and quaternary structures). Via their three-dimensional structure, cellular proteins are able to interact with each other and thereby dictate an organism’s physiology.
The primary structure of PrPC across different mammalian species is highly conserved (50) and is nearly identical to that of PrPSc (51). PrPSc and PrPC also share important posttranslational modifications, including the presence of two N-glycosylation sites, a single disulfide bond between cysteine residues, and a glycosylphosphatidylinositol membrane anchor at the C-terminus (this is the site of attachment of PrPC to the cell membrane) (52).
The biochemical and biophysical differences that exist between PrPC and PrPSc are the result of specific posttranslational modifications that affect the secondary, tertiary, and quaternary structures of these proteins. In this respect, the most important difference between PrPC and PrPSc relates to their response upon exposure to detergents and proteases. PrPC is soluble in nondenaturing detergents and degrades when exposed to proteases. PrPSc, on the other hand, forms amyloid structures (prion rods that show the typical fluorescence birefringence of amyloids) when exposed to detergents and is highly resistant to cellular proteases, particularly proteinase K (PK) (52). It is important to note that PrPSc is not fully resistant to PK, however, and this will be discussed further in the following texts. The insolubility of PrPSc to detergents, on the other hand, is universal, and this property is used in the laboratory to accurately diagnose the presence of prion disease.
Our knowledge of the structural differences that exist between PrPSc and PrPC at an atomic level has been obtained via careful studies on different transgenic animal models using nuclear magnetic resonance (NMR) and infrared (IR) spectroscopy and x-ray crystal diffraction. IR spectroscopy studies have shown that PrPC has a high alpha-helix content and virtually no beta-sheet content, whereas PrPSc is characterized by roughly equal amounts of alpha-helices and beta-sheets (53). Alpha-helices and beta-sheets are secondary protein structures. NMR spectroscopy studies also reveal that PrPC forms a unique three-dimensional structure in its C-terminus, consisting of three alpha-helices bundled with two short antiparallel beta-sheets (52), whereas the N-terminus does not arrange into a specific conformation. These findings have been largely replicated using x-ray crystal diffraction methods (54).
Possible Mechanisms of PrPC Conversion and PrPSc Propagation
The infectious nature of prions in animals was perhaps most influentially demonstrated by Legname and colleagues (55) in 2004, when they inoculated fibrils of N-terminally truncated recombinant mouse PrP made in Escherichia coli into the brains of transgenic PrP knockout mice expressing N-terminally truncated PrP and showed that these mice developed neurologic dysfunction between 380 and 660 days after inoculation, with neuropathologic findings consistent with prion disease. When brain extracts from these mice were then inoculated into the brains of wild-type mice, these mice developed a prion disease. Many other similar experiments have confirmed that prions have the ability to replicate and propagate within the nervous system.
As stated previously, the mode of propagation of prions is unique among infectious agents, in that prions do not appear to use nucleic acid to replicate within the host species. Instead, according to the protein-only hypothesis of prion spread, the presence of PrPSc by itself induces endogenous PrPC to misfold into PrPSc via the modification of the secondary and tertiary structures of PrPC in a process referred to as template-directed misfolding. In other words, PrPSc acts as a template that guides PrPC misfolding: one molecule of PrPSc induces the conversion of one molecule of PrPC into PrPSc, two molecules of PrPSc induce the transformation of two new molecules of PrPC, and so PrPSc accumulates in an exponential fashion (see Figs. 18.2 and 18.3. The progressive accumulation of pathogenic PrPSc in the nervous system leads to the clinical manifestations and neurodegeneration seen in prion disease (1).
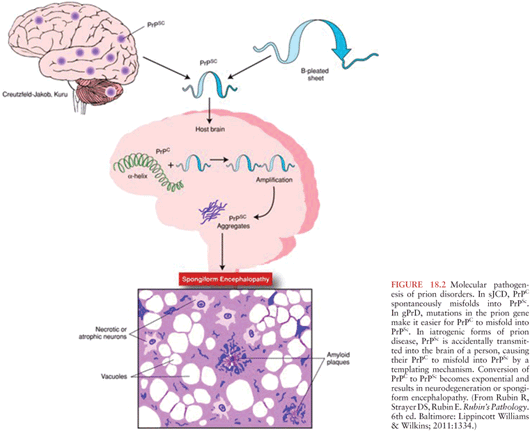
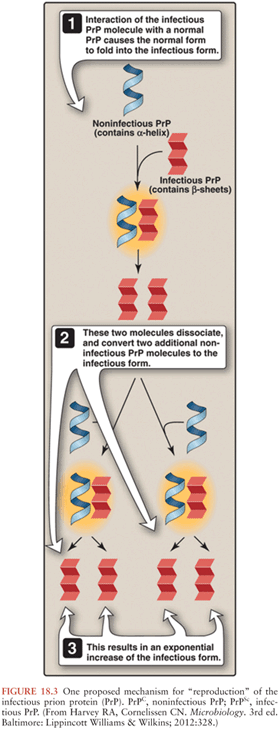
The exact mechanisms by which PrPSc leads to the misfolding of PrPC are yet to be determined. It has been demonstrated experimentally that the first step in prion infectivity involves the successful cellular uptake of PrPSc in susceptible host cells (56,57), thereby allowing the physical interaction between PrPC and PrPSc that is required for misfolding. Although it is widely accepted that the presence of cellular PrPC is required for the propagation of prion disease (58,59), the initial cellular uptake of PrPSc occurs without participation of PrPC (60). Instead, several cell membrane receptors have been implicated in the initial steps of transformation of PrPC into PrPSc. Among these, the low-density lipoprotein receptor–related protein 1 (LRP1) might be an important player (61) as well as glycosaminoglycan (62) and laminin receptors (63). There are likely other cellular receptors involved in this process.
Once incorporated to the host cell, PrPSc leads to PrPC misfolding within minutes. This was elegantly demonstrated in in vitro experiments using epitope-tagged PrPC in neuroblastoma cell lines (64). Moreover, it appears that PrPC misfolding occurs largely within the cell surface, from where PrPSc is then endocytosed and either recycled back to the plasma membrane or deposited within the Golgi system (65).
Several biochemical and biophysical factors have been shown to facilitate the misfolding of PrPC into PrPSc in vitro, including the presence of acidic and/or salt solutions and interactions between PrPC and metal ions (66). The cellular mechanisms of PrPC misfolding remain elusive, however, and there is mounting evidence suggesting that there are several different cellular structures and cofactors mediating the transformation of PrPC to PrPSc (67,68).
SPORADIC HUMAN PRION DISEASE: JAKOB-CREUTZFELDT DISEASE, FATAL INSOMNIA, AND VARIABLY PROTEASE-SENSITIVE PRIONOPATHY
Sporadic Jakob-Creutzfeldt Disease
The disease that we now recognize as sporadic Jakob-Creutzfeldt disease (sJCD) clinically and neuropathologically was first described by Alfons Maria Jakob (69,70) in the early 1920s. The single case that his mentor Hans Gerhard Creutzfeldt (71) described in 1920, on the other hand, was neither clinically nor neuropathologically representative of sJCD or what we now know as prion disease (72,73). Therefore, some authors prefer the more correct eponym Jakob’s or Jakob-Creutzfeldt disease rather than Creutzfeldt-Jakob disease (CJD) when referring to prion disease. The authors of this chapter prefer the name Jakob-Creutzfeldt over Creutzfeldt-Jakob. Thus, the term should probably be Jakob-Creutzfeldt disease (or even Jakob’s disease [JD]). Of note, some physicians have mistakenly thought prion disease was due to the JC virus and incorrectly sent the JC virus PCR as a test for prion disease. In this chapter, we use the term Jakob-Creutzfeldt disease.
Most studies have shown that about 85% of human prion diseases are sporadic, 10% to 15% are genetic, and less than 1% is acquired. In a large prospective systematic study of human prion disease in Europe, Australia, and Canada from 1993 to 2002, which included more than 4,400 pathology-proven cases, the incidence of sporadic prion disease was approximately 85%, genetic was about 10%, and acquired were about 6% (74). The larger percentage of acquired cases than many other studies is likely due to the emergence of vJCD primarily in the United Kingdom and France during this time period (75) as well as the emergence of human pituitary hormone cases from prior exposure in the United Kingdom, France, and Australia (76). The incidence of sporadic human prion diseases in most populations is about 1 per million, of gPrD about 1 per 10 million, and even less for acquired prion disease (77).
Before recent advances in molecular biology were applied to study human prion disease, sJCD was subdivided into separate phenotypes based on clinical and histopathologic characteristics. Classic subtypes of sJCD described in the early literature include the Heidenhain variant of sJCD, characterized by prominent early visuospatial symptomatology, sometimes leading to cortical blindness (78); the Jakob variant or spastic pseudosclerosis, which presents with prominent frontal, pyramidal, and extrapyramidal symptoms; and the Brownell-Oppenheimer variant, characterized by early cerebellar dysfunction and late dementia. Other clinical variants exist in the literature.
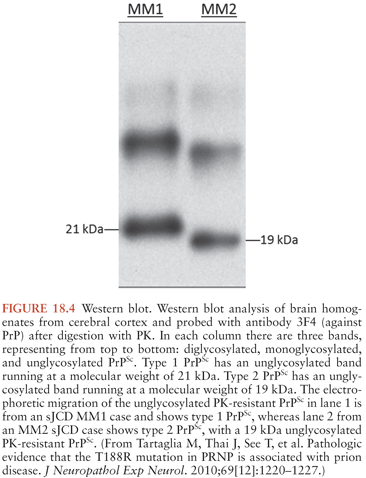
It is now known that the clinicopathologic variability of sJCD is in part due to two factors: the polymorphisms at codon 129 of PRNP (36) and at least two distinct “species” of PrPSc (PrPSc type 1 and type 2). In the laboratory, the two types of PrPSc are distinguished by their mobility on Western blot transfers after exposure to PK (Fig. 18.4) (79). Taken together, this line of evidence suggests that the different sJCD phenotypes are an expression of interactions of the host PRNP genotype and PrPSc type, and this notion forms the basis of our current classification of sJCD.
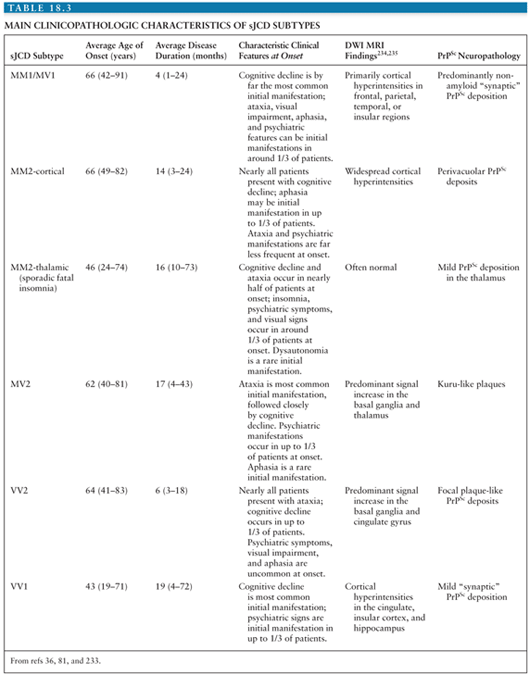
Large studies have been carried out comparing molecular, neuropathologic, and clinical characteristics of sJCD subjects (80). Results of these studies prompted a classification scheme that included six molecular subtypes based on PRNP codon 129 polymorphisms (M/M, M/V, or V/V) and PrPSc type (type 1 or 2). There is considerable overlap between classic sJCD clinical phenotypes and the clinical phenotypes associated with each molecular subtype to the extent that five combined sJCD subtypes were initially recognized: MM1/MV1, VV1, VV2, MV2, and MM2 cortical and MM2 thalamic (the latter of which corresponds to sporadic fatal insomnia) (79,81). The discovery that many patients have both type 1 and type 2 prions has complicated this earlier molecular classification and expanded the subtypes of sJCD (79,82). It is beyond the scope of this chapter to describe the clinicopathologic characteristics of each subtype in great detail. The main characteristics of each subtype are shown in Table 18.3 (79).
The clinical presentation of sJCD in general is protean, however. Studies show that 40% to 60% of affected individuals initially develop cognitive symptoms (83–85), generally manifesting as memory, executive, and language dysfunction and/or confusion. Other early clinical manifestations include cerebellar dysfunction, constitutional symptoms, and behavioral/personality changes (e.g., depression, irritability) in around 20% of pathology-proven cases of sJCD. Furthermore, impaired vision (blurred vision, diplopia, and visual hallucinations) has been reported as an initial complaint in 9% to 15% of cases (84–86). Myoclonus, which is anecdotally considered to be a sensitive sign of sJCD by many clinicians, is a late feature of the disease in up to 90% of cases (84). Other movement disorders are often seen in patients with sJCD, either early or late in their disease course, including dystonia (fixed posturing of a body part), choreoathetosis (slow, writhing or quick dance-like movements), parkinsonism (slowed movements, tremor at rest, etc.), and supranuclear gaze palsy (inability to move eyes volitionally) (87). Other rare initial manifestations of sJCD include seizures (including status epilepticus) (88); these have been reported in some studies in up to 10% of cases, but usually later in the disease course. In our experience, seizures are far less common than this (84,86,88). Clinically evident peripheral neuropathy (including cranial neuropathy) is very uncommon in sJCD (89,90) but is seen more commonly in some gPrDs (91–95).
The average age of onset of sJCD is 65 years, with a peak incidence in the 7th decade. The average disease duration is 4 to 7 months. The great majority of patients (90%) die within 1 year of diagnosis, but up to 5% of patients may survive for 2 or more years (36,84,96). Certain factors such as younger age at onset, female gender, and heterozygosity at codon 129 have been associated with longer survival in sJCD (36,77,79). Invariably, however, sJCD leads to a relatively rapidly progressive dementia (RPD) (compared to most other neurodegenerative diseases), with varied clinical manifestations, depending on which areas of the brain are affected by prion neurodegeneration. Regardless of pattern of prion spread, most cases develop akinetic mutism prior to death (87). Death is usually due to aspiration pneumonia.
Brain magnetic resonance imaging (MRI) is the most sensitive and specific diagnostic test for sJCD, with published sensitivity of approximately 91% to 96% and specificity of approximately 92% to 94% (79,97,98). The high diagnostic sensitivity of brain MRI is largely a reflection of its ability to detect changes very early in the disease course (98). The most sensitive MRI sequences for the diagnosis of sJCD are diffusion-weighted imaging (DWI) and apparent diffusion coefficient (ADC) sequences; indeed, DWI has been shown to be more sensitive than fluid-attenuated inversion recovery (FLAIR)/T2 sequences abnormalities (98,99).
The radiographic hallmark of sJCD is the presence of DWI MRI hyperintensities in the neocortex, basal ganglia, and/or thalamus, often associated with corresponding ADC map hypointensities, suggesting restricted flow of water molecules in these regions (97), probably due to pathological changes occuring in prion disease, particularly vacuolation (spongiform change), prion deposition, and/or gliosis (astrocyte proliferation) (100). These MRI findings have been included in various criteria for sJCD (101), although some criteria allow T2 or FLAIR and do not require DWI abnormalities (102), which might lead to false-positive diagnoses (97). An example of the typical brain MRI findings seen in sJCD is shown in Figure 18.5. Three major patterns of DWI MRI hyperintensities have been identified in sJCD: neocortical and subcortical (approximately 68% of cases), predominantly neocortical (24% to 30%), and predominantly subcortical (mostly striatal hyperintensities, with or without thalamic involvement) (2% to 5%) (97,103) (Fig. 18.6). Cortical hyperintensities (or cortical ribboning) can be unilateral or bilateral but typically spares the precentral cortex (97,104). Striatal involvement often begins unilaterally, but eventually becomes bilateral, and typically has a decreasing anterior-posterior gradient (i.e., the anterior caudate appears more hyperintense than the posterior putamen) (97). Moreover, up to 90% of sJCD cases demonstrate involvement of limbic and paralimbic cortical structures (i.e., insula, anterior cingulate, hippocampus), visualized as hyperintensities and hypointensities of these regions on DWI and ADC sequences, respectively. In our experience, isolated limbic involvement is atypical for sJCD, however, and should alert the clinician toward alternative diagnoses (97).
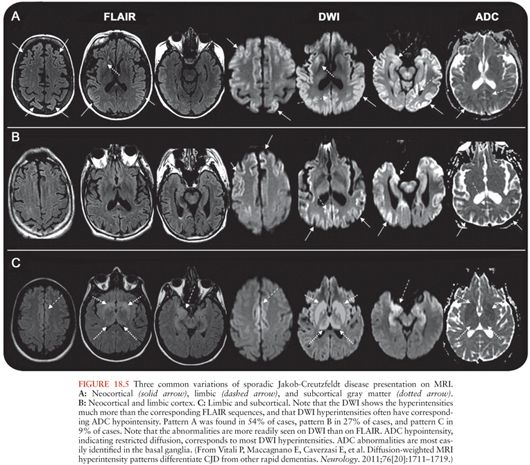
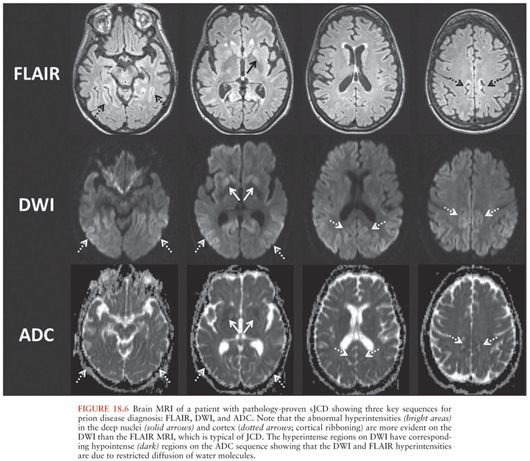
Cortical ribboning is not 100% sensitive and specific for sJCD, however, as it is also seen in cases of viral and/or autoimmune encephalitis, hypoglycemia, seizures and status epilepticus, hyperammonemic encephalopathy, hypoxic injury, mitochondrial encephalopathy, vasculitis, and other conditions (97,98,105). Hence, clinicians should consider these and other conditions in the presence of cortical ribboning on brain MRI. Likewise, striatal and diencephalic DWI hyperintensities with ADC hypointensities should not be considered pathognomonic of sJCD, as these changes have been observed in extrapontine myelinolysis, Wilson disease, Wernicke encephalopathy, and hyperglycemia with seizures (97,105). These DWI hyperintensities might decrease in late stages of the disease due to brain atrophy, particularly in patients with disease duration of over 1 year (97). Despite the high sensitivity and specificity of MRI DWI and ADC sequences for the diagnosis of sJCD in the right clinical setting, there is evidence to suggest that radiologists often miss the diagnosis of sJCD because they fail to recognize these changes (106,107).
Electroencephalogram
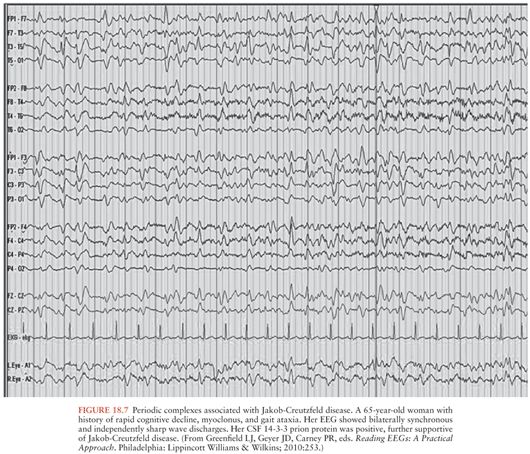
The diagnostic value of electroencephalography (EEG) has lessened to some extent due to the high sensitivity and specificity of brain MRI for the diagnosis of sJCD (98). Still, the presence of periodic sharp wave complexes (PSWCs), often biphasic or triphasic, occurring every 0.5 to 2 seconds (Fig. 18.7) remains a useful finding that helps rule in sJCD in the right clinical setting (77,108). This EEG finding is found only in about two thirds of sJCD patients, however, and is most often present late in the disease course and may require repeated EEG testing (108). In general, compared to brain MRI, EEG probably provides little value for diagnosis early in the disease course. Furthermore, the presence of PSWCs rarely is seen in other neurologic conditions such as Alzheimer disease, dementia with Lewy bodies, toxic-metabolic and anoxic encephalopathies (e.g., hepatic), progressive multifocal leukoencephalopathy, and Hashimoto encephalopathy (109,110).
Cerebrospinal fluid
Routine CSF analysis is typically normal in sJCD, although sometimes a mild elevation in the total protein level can be seen (typically less than 75 mg/dL). CSF pleocytosis (>10 cells/µL white blood cells [WBCs]), an elevated immunoglobulin G (IgG) index, and/or the presence of oligoclonal bands are unusual in sJCD and should lead the clinician to consider other conditions, particularly infectious or autoimmune disorders (111,112).
The clinical use of several CSF biomarkers is quite controversial, with evidence showing varying degrees of sensitivity and specificity for the diagnosis of sJCD. The 14-3-3 protein, for example, which is one of the first CSF proteins reported to be elevated in sJCD, and initially reported to have 100% sensitivity and 96% specificity for the diagnosis of sJCD (113), has been subsequently found to have more limited sensitivity and specificity when studied in larger patient cohorts (114,115). A more recent analysis in the United Kingdom showed CSF 14-3-3 protein sensitivity of 86% and specificity of 74% in a pathologically confirmed cohort (116). Similarly, an analysis of data obtained from the U.S. National Prion Disease Pathology Surveillance Center (NPDPSC) showed the 14-3-3 protein Western blot only had a receiver operating characteristic area under the curve (ROC AUC) value of 0.68 (117), which is poor considering that a test with perfect sensitivity and specificity would be 1.0. Some studies on the clinical use of CSF 14-3-3 protein suggests it should be considered a general marker of neuronal injury and death. Indeed, 14-3-3 protein is ubiquitously present in the intracellular compartment of brain neurons and is released into the CSF compartment due to neuronal injury from varied causes. Thus, 14-3-3 protein can be elevated in many non-prion neurologic conditions, such as multiple sclerosis, acute stroke, Alzheimer disease and other neurodegenerative dementias, HIV-related neurodegeneration, seizures, and many others (118–122). In this respect, a large multicenter European study found that the specificity of 14-3-3 protein is lowered when used to differentiate sJCD from acute neurologic disorders (such as vascular, inflammatory, or seizures) versus neurodegenerative dementias (82% to 87% vs. 95% to 97% respectively). This suggests taking the differential diagnoses into consideration is important when interpreting the test result (123). In summary, a positive 14-3-3 protein does not necessarily equate with prion disease. Regardless, recent recommendations from the American Academy of Neurology suggest ordering CSF 14-3-3 protein when there is a strong clinical suspicion of JCD (pretest probability of 20% to 90%) in order to reduce diagnostic uncertainty (124).
Total-tau (t-tau), neuron-specific enolase (NSE), and the astrocytic protein S100β are also used as CSF biomarkers for sJCD diagnosis. The sensitivity and specificity of these biomarkers varies greatly among studies. In a large multicenter, retrospective European study (115), the combined sensitivity and specificity of the four main sJCD biomarkers (14-3-3 protein, t-tau, NSE, and S100β) was found to be higher than the individual sensitivity and specificity. Not all patients in this study underwent all four tests nor were the tests performed in the same CSF samples, however; hence, this study did not permit a fair comparison between biomarkers. Nevertheless, the study found the sensitivity and specificity of the 14-3-3 protein to be 85% and 84%, t-tau (cutoff >1,300 pg/mL) 86% and 88%, NSE 73% and 95%, and S100β 82% and 76%, respectively (115). Other studies, however, have shown sensitivity and specificity values higher than 90% for t-tau in sJCD (125,126), although there is still no agreement over its cutoff value (usually higher than 1,150 pg/mL).
In reviewing all of the data, the opinion of the authors of this chapter is that among 14-3-3, t-tau and NSE, the single best CSF biomarker is the t-tau, although NSE has somewhat higher specificity, whereas the 14-3-3 protein is overall the least clinically useful. There probably is not sufficient data to make a determination of the clinical utility of S100B (beta). At our center, we send all three biomarkers when looking for evidence of rapid neuronal injury. In summary, these CSF biomarkers are likely markers of rapid neuronal injury and do not equate with prion disease, but might be helpful in some cases, yet have overall less diagnostic use than brain MRI (97,98). New CSF diagnostic tests, such as real-time quaking-induced conversion reaction (RT-QuIC), that are relatively specific for prions (and not just biomarkers) are under development (127).
Two Uncommon Forms of Sporadic Human Prion Disease: Fatal Insomnia and Variably Protease-Sensitive Prionopathy
The MM2 thalamic form of sJCD is sometimes referred to as sporadic fatal insomnia (sFI), the least common of the six forms of sJCD based on molecular classification (36,81). It is similar clinically and pathologically to the rare gPrD FFI, as described under gPrDs section. It is the rarest form of sporadic human prion disease, with approximately 31 cases reported in the literature. On average, affected patients are 46 years old at disease onset, mean survival is 24 months (79). Clinicians should consider this form of sporadic prion disease in patients with rapidly progressing dementia associated with sleep abnormalities and dysautonomia. Unlike sJCD, EEG, CSF, and brain MRI investigations have not been shown to be helpful for the diagnosis of this disorder (79). Instead, polysomnography, which shows impaired sleep-related wave forms (such as K-complexes and spindles), and brain positron emission tomography (PET) examinations, which consistently show thalamic hypometabolism, have proven to be more useful diagnostic tools (79).
The variably protease-sensitive prionopathy (VPSPr) is the latest addition to the list of sporadic human prion diseases introduced into the literature in 2008 (128). The term variably protease-sensitive prionopathy is derived from the fact that prions extracted from brain tissue from such cases produces a distinctive electrophoretic profile on Western blot due to sensitivity to protease digestion, which distinguishes it from other forms of prion diseases. As most laboratory techniques for diagnosis of prion disease (e.g., Western blot and immunohistochemistry) rely on identification of protease-resistant PrPSc, as described previously, detecting this form of prion disease can be quite difficult (129). VPSPr has certain distinctive clinical, molecular, and pathologic characteristics (79,129). Clinically, patients generally present with prominent psychiatric manifestations and rapid cognitive decline (primarily aphasia), followed by motor manifestations (ataxia and parkinsonism). Clinical presentations, however, vary according to codon 129 genotype (79). Based on the cases identified through 2010, cases associated with methionine homozygosity (VPSPr-129MM) seem to not have prominent psychiatric features, whereas VPSPr-129MV and VSPr-129VV cases usually lack parkinsonism and ataxia. As is the case in sJCD MM2-thalamic form, MRI, CSF, and EEG investigations often do not show the typical features diagnostic for sJCD. It is not clear if VPSPr is as transmissible as other prion diseases, as the prions in VPSPr are more sensitive to proteases and thus might be more easily degraded.
Basic Neuropathology of Human Prion Diseases
At a macroscopic level, atrophy is the only gross neuroanatomic finding in sJCD brains. Atrophy can be mild to moderate, localized to the cerebral cortex, basal ganglia, and cerebellar regions. As in other neurodegenerative diseases, atrophy is most pronounced in patients with a long clinical course. Unlike what is seen in other neurodegenerative diseases, however, prominent hippocampal atrophy is usually not seen in human prion disease (130).
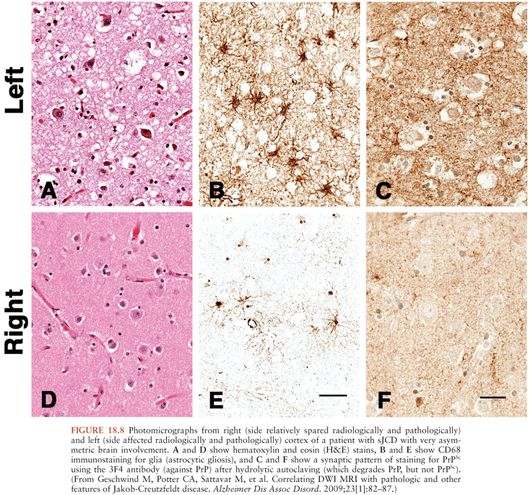
Microscopically, the histopathologic hallmarks of human prion disease are the presence of neuronal loss, astrocytic gliosis, vacuolation (or spongiform changes), and deposition of prions (PrPSc) in the brain (Fig. 18.8) (131). These changes occur without signs of inflammation in the brain, other than those seen typically with neuronal loss in neurodegenerative disease, such as microglia activation, astrocytosis, and release of cytokines (132–134). The distribution of vacuolation changes seen in human prion disease vary between cases and disease subtypes (77), although the head of the caudate nucleus is most often involved, whereas the brainstem is relatively spared (130). More than 95% of sJCD cases show vacuolation and the remaining cases are diagnosed by immunohistochemistry to PrPSc. PrPSc deposition in sJCD is usually in a nonamyloid form, with frequent “synaptic” or punctate deposition in the cortex as well as perivacuolar PrPSc deposition in brain regions containing vacuolation.
Related posts:
 Neurologic Manifestations of Varicella and Herpes Zoster
Neurologic Manifestations of Varicella and Herpes Zoster
 Meningitis and Encephalitis Caused by Mumps Virus
Meningitis and Encephalitis Caused by Mumps Virus
 Human Immunodeficiency Virus
Human Immunodeficiency Virus
 Vaccines for Viral Diseases with Significant Central Nervous System Manifestations
Vaccines for Viral Diseases with Significant Central Nervous System Manifestations
 Subdural Empyema and Suppurative Intracranial Phlebitis
Subdural Empyema and Suppurative Intracranial Phlebitis
 Rickettsioses, Anaplasmoses, and Q Fever
Rickettsioses, Anaplasmoses, and Q Fever
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


