Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Patrick G. Gallagher
Bertil Glader
This chapter focuses on hemolytic disorders and abnormalities of red blood cell (RBC) shape resulting from alterations of the erythrocyte membrane. The major emphasis is on the hereditary spherocytosis (HS) and hereditary elliptocytosis (HE) syndromes because these are most commonly encountered by clinicians.1 The traditional classification of these disorders, retained in this chapter because of its clinical applicability (Fig. 27.1), has been based on RBC shape changes. There has been an explosion of knowledge regarding the biology of the erythrocyte membrane, providing a better understanding of membrane structure and function as well as revealing the heterogeneous and diverse pathobiology of these disorders. Readers should consult prior editions of this text for details of previously cited primary references.
THE ERYTHROCYTE MEMBRANE
The erythrocyte membrane, because of its easy accessibility, is the most studied biologic membrane. Composed of a lipid bilayer and an underlying cortical membrane skeleton (Fig. 27.2), the erythrocyte membrane provides the red cell with the deformability and stability required to withstand its travels through the circulation. The erythrocyte is subjected to high sheer stress in the arterial system, dramatic changes in size in the microcirculation with capillary diameters as small as 7.5 µm, and marked fluctuations in tonicity, pH, and pO2 as it travels through the body.2
The lipid bilayer is composed primarily of phospholipids intercalated with unesterified cholesterol and glycolipids. Phospholipids are asymmetrically organized, with the choline phospholipids, phosphatidylcholine (PC) and sphingomyelin (SM), primarily in the outer half of the bilayer; and the amino phospholipids, phosphatidylethanolamine (PE) and phosphatidylserine (PS), in the inner half of the bilayer.3 In pathologic states, such as thalassemia, sickle cell disease, and diabetes, loss of phospholipid asymmetry with externalization of PS leads to activation of blood clotting via conversion of prothrombin to thrombin and facilitates macrophage attachment to erythrocytes, marking them for destruction.4
Lipids are arranged in domains within the bilayers, as large, lipid-rich macroscopic domains, protein-bound microscopic domains, or domains associated with the detergent-resistant fraction of the membrane (called DRM or lipid rafts).5, 6 The role(s) of these domains has yet to be elucidated, but DRM domains may facilitate malarial invasion of the erythrocyte.7, 8
The erythrocyte membrane contains about 15 major proteins and hundreds of minor ones.9, 10, 11 One erythrocyte proteomic study found 340 of 527 proteins were membrane-associated.12 A human erythrocyte proteome database is maintained at the Max-Planck Unified (MAPU) proteome database: http://www.mapuproteome.com/rbc.13 Typically, membrane proteins are classified as integral, penetrating or crossing the lipid bilayer and interacting with the hydrophobic lipid core, or peripheral, interacting with integral proteins or lipids at the membrane surface but not penetrating into the bilayer core. The integral membrane proteins include the glycophorins, the Rh proteins, Kell and Duffy antigens, and transport proteins such as band 3 (AE1, anion exchanger 1), Na+,K+-ATPase, Ca2+-ATPase, and Mg2+-ATPase. An assortment of membrane receptors and antigens are present on various integral membrane proteins. Peripheral membrane proteins include the structural proteins of the spectrin-actinbased membrane skeleton.14
The membrane skeleton, composed of an intricately interwoven meshwork of proteins, interacts with both integral membrane proteins and the lipid bilayer.14 The major proteins of the membrane skeleton include spectrin, actin, ankyrin, protein 4.1R, and protein 4.2 (Fig. 27.2 and Table 27.1).
Spectrin is the principal structural component of the membrane skeleton, as well as its most abundant protein, comprising 25% to 30% of membrane protein. Spectrin functions include provision of structural support for the lipid bilayer, maintenance of cellular shape, and regulation of lateral movement of integral membrane proteins.9 Spectrin is composed of α and β chains, which, despite a number of similarities, including both containing 106 amino acid α-helical “spectrin repeats” composed of triple-helices linked by short connecting regions, are distinct proteins encoded by separate genes. αβ-Spectrin chains intertwine in an antiparallel manner to form 100-nm long heterodimers. These αβ-spectrin heterodimers self-associate with other αβ-spectrin heterodimers to form (αβ)2 heterotetramers, the functional spectrin subunit in the erythrocyte.15 Tetramers provide significant flexibility and structural support for the lipid bilayer, helping maintain cellular shape.9, 16 Spectrin heterodimer-tetramer interconversion is governed by a simple thermodynamic equilibrium that favors tetramer formation.17
The primary linkage of spectrin to the plasma membrane is via binding of spectrin tetramers to ankyrin, which in turn binds to the integral protein band 3.18 Protein 4.2 binds to band 3 and probably to ankyrin, presumably promoting their interactions.19
A second linkage of spectrin to the plasma membrane is mediated by its association with the “junctional complex,” a multiprotein complex that includes spectrin, actin, adducin, and protein 4.1R.20 Protein 4.1R directly interacts with both spectrin and actin, as well as other proteins of the junctional complex and the plasma membrane including band 3, glycophorin C (GPC), p55, calmodulin, CD44, pIC1n, CASK, mature parasite-infected antigen, and phosphatidylserine. Other important junctional complex proteins include the β subtype of actin, tropomyosin, tropomodulin, p55, and adducin.9 Adducin is a heterodimer of structurally related proteins, α-, β-, or γ-adducin, which functions in the early assembly of the spectrin/actin complex by capping the ends of fast-growing actin filaments and by recruiting spectrin to the ends of actin filaments.20, 21
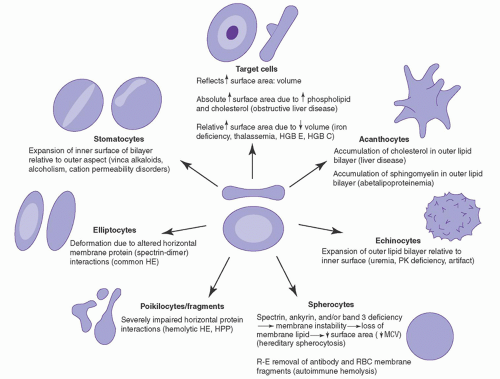
FIGURE 27.1. Diagrammatic representation of abnormal cells associated with alterations in the erythrocyte membrane. HE, hereditary elliptocytosis; HGB, hemoglobin; HPP, hereditary pyropoikilocytosis; MCV, mean corpuscular volume; PK, pyruvate kinase; RBC, red blood cell; R-E, reticuloendothelial.
Another membrane skeleton linkage to the plasma membrane is mediated via binding of a multiprotein complex containing the Rh proteins, the RH-associated glycoproteins, CD47, LW, glycophorin B, and protein 4.2 to ankyrin.
Erythrocyte membrane disorders result from alterations in the quantity or quality (or both) of individual proteins and their dynamic interactions with each other.2, 22 Disruption of the vertical protein-protein interactions of the membrane, i.e., the spectrin-ankyrin-band 3 linkage or the band 3-protein 4.2 interaction, leads to uncoupling of the membrane skeleton from the lipid bilayer. This leads to membrane instability with loss of lipids and some integral membrane proteins, resulting in loss of membrane surface area and the phenotype of spherocytosis (Fig. 27.2). Disruption of the horizontal interactions of membrane skeleton proteins, including perturbation of spectrin self-association or junctional complex protein-protein interactions, leads to membrane instability, altered membrane deformability and mechanical properties, and the phenotype of elliptocytosis (Fig. 27.2).23
Development of biochemical techniques has allowed the separation and quantification of membrane proteins (Fig. 27.3) and the detection of membrane protein abnormalities in many hereditary red cell disorders. Advances in molecular biology have allowed determination of the precise genetic defects in many cases.
HEREDITARY SPHEROCYTOSIS
HS is a hemolytic disorder characterized by anemia, intermittent jaundice, splenomegaly, and responsiveness to splenectomy.24, 25 There is marked clinical heterogeneity, ranging from an asymptomatic condition to a fulminant hemolytic anemia. The morphologic hallmark of HS is the spherocyte, created by loss of membrane surface area and characterized by abnormal erythrocyte osmotic fragility (OF) in vitro. The interesting history of HS is recounted in detail by Dacie26 and by Packman.27 The earliest clinical account of the disorder is probably the 1871 report of Vanlair and Masius,28 and the 1934 studies of Haden drew attention to a probable structural abnormality of the membrane as the basis for hemolysis.29 Subsequent investigation of HS afforded important insights into the structure and function of cell membranes and the role of the spleen in maintaining erythrocyte integrity.
Prevalence and Genetics
HS is the most common of the hereditary hemolytic anemias among people of northern European descent. In the United States, the incidence of the disorder is approximately 1 in 3,000 to 5,000.24, 25 When mild and asymptomatic forms of the disease are considered, the incidence is closer to 1:2,000.30, 31 The disease is encountered worldwide, but its incidence and prevalence in other ethnic groups are not clearly established.
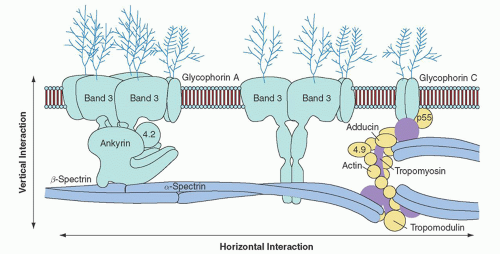
FIGURE 27.2. The erythrocyte membrane. A model of the major proteins of the erythrocyte membrane is shown: α– and β-spectrin, ankyrin, band 3 (the anion exchanger), 4.1R (protein 4.1R) and 4.2 (protein 4.2), Rh (Rhesus polypeptide), RhAG (Rh-associated glycoprotein), LW (Landsteiner-Weiner glycoprotein), actin, and glycophorin. Membrane protein-protein and protein-lipid interactions are often divided into two categories: (1) vertical interactions, which are perpendicular to the plane of the membrane and involve spectrin-ankyrin-band 3-protein 4.2 interactions and weak interactions between spectrin and the negatively charged lipids of the inner half of the membrane lipid bilayer; and (2) horizontal interactions, which are parallel to the plane of the membrane and include interactions between junctional complex proteins and spectrin or other membrane proteins. (Reprinted with permission from Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet 2008;372:1411.)
In the majority of affected families, HS is transmitted in an autosomal dominant manner. Up to 25% of HS patients demonstrate nondominant inheritance, and the parents of these patients are clinically and hematologically normal.32, 33 In many of these patients, HS is due to a de novo mutation. In others, it is inherited in an autosomal recessive fashion, typically linked to abnormalities in the α-spectrin or protein 4.2 genes. A few cases of severe, homozygous HS have been reported, usually in cases where there is parental consanguinity.34, 35
Pathogenesis
The pathogenesis of HS involves the interplay between an intrinsic erythrocyte membrane protein defect and an intact spleen that selectively retains, damages, and eventually destroys abnormal HS erythrocytes.
Membrane Protein Defects
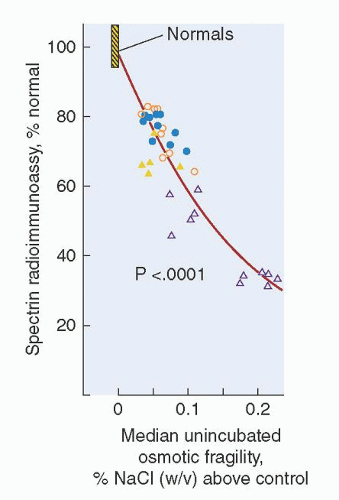
The primary lesion in HS erythrocytes is caused by membrane protein defects that result in membrane instability. The first biochemical defect recognized in patients with HS was spectrin deficiency, and the degree of spectrin deficiency was found to correlate with the extent of spherocytosis, the degree of abnormality of the OF test, and the severity of hemolysis (Fig. 27.4).32, 33, 36, 37, 38 In some cases, spectrin deficiency is the result of impaired synthesis, whereas in other instances, it is caused by quantitative or qualitative deficiencies of other proteins that integrate spectrin into the cell membrane, especially ankyrin. In the latter case, free spectrin is degraded, thereby leading to spectrin deficiency. Analysis of red cell membrane proteins in patients with HS has identified several major abnormal patterns: spectrin deficiency alone, combined spectrin and ankyrin deficiency, band 3 deficiency, protein 4.2 deficiency, or no obvious biochemical abnormality. Each of the variant subsets is associated with mutations that result in different protein abnormalities and varied clinical expression (Table 27.2).39 A tabulation of these mutations, searchable by gene or phenotype, maintained by investigators from Yale University and the National Human Genome Research Institute of the National Institutes of Health, is available at http://research.nhgri.nih.gov/RBCmembrane/.
Spectrin Deficiency
In the erythrocyte, α-spectrin is synthesized in three- to four fold excess with β-spectrin synthesis rate-limiting for membrane assembly of αβ-spectrin heterotetramers.40, 41 Excess α-chains normally are degraded. Clinical abnormalities caused by α-spectrin deficiency are found only in the homozygous or compound heterozygous state, as heterozygotes produce sufficient normal α-spectrin to balance β-spectrin production and maintain a normal phenotype. In contrast, defects of β-spectrin are clinically apparent in the heterozygous state because synthesis of β-spectrin is rate-limiting. Red cell membranes from patients with autosomal recessive HS due to α-spectrin deficiency have only 40% to 50% the normal amount of spectrin, whereas spectrin levels range from 60% to 80% in patients with autosomal dominant HS due to β-spectrin defects.24, 25
Determination of the precise genetic defects in α-spectrin-linked HS has been slow. The large size of the α-spectrin gene (52 exons) and decreased mRNA associated with presumed null alleles has made mutation detection difficult. However, several variant alleles have been associated with α-spectrin-deficient, nondominant HS.42, 43 These include spectrinBug Hill, an alanine to aspartic acid substitution in the αII domain, and spectrinLEPRA, low-expression Prague, a C to T substitution associated with increased utilization of an alternative acceptor splice site in intron 30. Whether these are disease-causing variants or are in linkage disequilibrium with other, as yet uncharacterized mutations, has not been determined.
Mutations of β-spectrin impair β-spectrin synthesis or produce truncated β-spectrin chains that are unstable or do not bind to ankyrin and, thus, are not assembled onto the membrane skeleton.44, 45 One HS-associated β-spectrin variant, β-SpectrinKissimmee, is a point mutation that leads to defective binding of spectrin to protein 4.1R.46 Occasionally, cases of β-spectrin-linked HS arise de novo.47
Ankyrin Defects
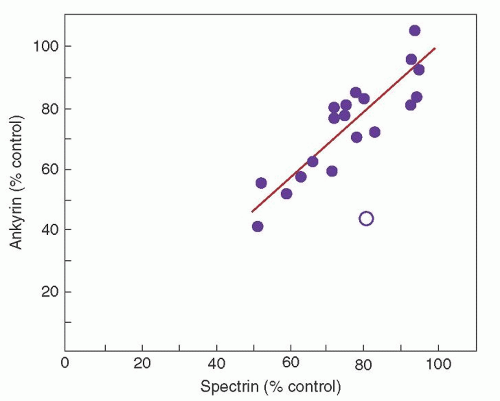
Combined spectrin and ankyrin deficiency is the most common biochemical abnormality found in the red cell membrane in cases of typical, autosomal dominant HS.48 Ankyrin is the principal binding site for spectrin on the red cell membrane, and studies of membrane skeleton assembly indicate that ankyrin deficiency leads to decreased incorporation of spectrin on the membrane despite normal spectrin synthesis. As expected, there is proportional deficiency of spectrin and ankyrin in these erythrocytes (Fig. 27.5).
TABLE 27.1 MAJOR HUMAN ERYTHROCYTE MEMBRANE PROTEINS, THEIR GENES, AND ASSOCIATED DISORDERS
aMultiple ankyrin isoforms are seen on SDS-PAGE gels numbered 2.1, 2.1, 2.3, etc.
bProtein 4.1R is a doublet (4.1a and 4.1b) on SDS-PAGE gels, with 4.1a derived from 4.1b by deamidation.
cGlycophorins are visible only on PAS-stained gels.
Modified with permission from Gallagher PG, Lux SE, Disorders of the erythrocyte membrane. In: Nathan DG, Orkin SH, Ginsburg D, Look AT, eds. Hematology of infancy and childhood, 6th ed. Philadelphia, PA: WB Saunders, 2003:567-568.
Ankyrin gene mutations are the most common defect associated with dominant HS. The majority of ankyrin defects are private point mutations in the coding region of the ankyrin gene associated with reduced or absent expression of the mutant allele.34 Around 15% to 20% of these mutations arise de novo.49 A few cases of spectrin/ankyrin-deficient HS are associated with mutations in the ankyrin gene erythroid promoter.50, 51, 52
HS has been associated with karyotypic abnormalities involving deletions or translocations of the ankyrin-1 gene locus on chromosome 8p. Ankyrin deletions may be part of a contiguous gene syndrome associated with HS, mental retardation, typical facies, and hypogonadism.53, 54
Band 3 Deficiency
Heterozygous band 3 mutations are found in 10% to 25% of patients with autosomal dominant HS.55, 56 Erythrocyte membranes demonstrate a 20% to 40% decrease of band 3 content and decreased or absent protein 4.2. Red cell anion transport is decreased proportional to the band 3 deficiency. Numerous band 3 gene mutations spread throughout the regions encoding both the cytoplasmic and membrane-spanning domains have been described. Many of these are null mutations associated with markedly reduced or absent band 3 mRNA. Others are missense mutations that disrupt band 3-membrane protein interactions or disrupt normal band 3 cellular trafficking from the endoplasmic reticulum to the plasma membrane.56, 57
Affected patients typically exhibit mild to moderate HS.24, 25 Mushroom-shaped or “pincered” red cells on peripheral smear have been associated with band 3-deficient HS. Homozygotes or compound heterozygotes for band 3 mutations usually exhibit more severe HS. Band 3 alleles that influence band 3 expression have been reported that, when inherited in trans to a band 3 mutation, aggravate band 3 deficiency and worsen the clinical phenotype.58, 59, 60 Although band 3 is expressed in the distal tubules of the kidney, only a few patients with band 3-associated HS exhibit defects in urinary acidification.61, 62
Protein 4.2 Deficiency
Autosomal recessive HS has been described in a subset of patients, primarily those of Japanese ancestry, whose erythrocyte membranes demonstrate marked deficiency of protein 4.2.63 In some cases, there is co-existing deficiency of band 3 and ankyrin. Clinical severity and red cell morphology are variable, with spherocytes, elliptocytes, or sphero-ovalocytes on peripheral smear. A few mutations of the protein 4.2 gene have been described in these patients, primarily null or missense mutations. One missense mutation, Protein 4.2Nippon, associated with a spherocytic, ovalocytic, and elliptocytic hemolytic anemia, is relatively common in Japan.64 Protein 4.2 deficiency also occurs in association with HS-associated mutations in the cytoplasmic domain of band 3, likely due to disruption of band 3-protein 4.2 interactions.
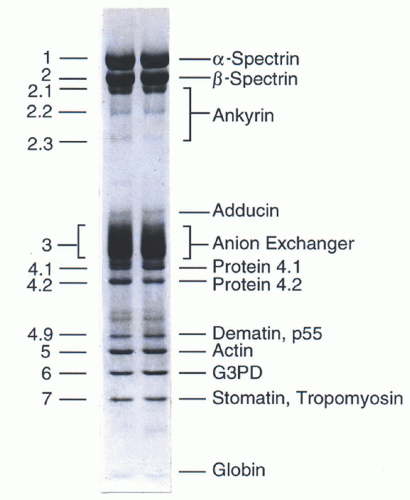
FIGURE 27.3. Protein composition of the red blood cell membrane skeleton. The major components of the erythrocyte membrane as separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and revealed by Coomassie blue staining. G3PD, glucose 3-phosphate dehydrogenase. (Reprinted with permission from Gallagher PG, Tse WT, Forget BG. Clinical and molecular aspects of disorders of the erythrocyte membrane skeleton. Semin Perinatol 1990;14:352.)
FIGURE 27.4. Relationship between spectrin deficiency and unincubated osmotic fragility in hereditary spherocytosis. Spectrin content, measured by radioimmunoassay, is shown on the vertical axis; and osmotic fragility, measured by the NaCl concentration that produces 50% hemolysis of red blood cells, is shown on the horizontal axis. Circles represent patients with typical, autosomal dominant HS and triangles represent patients with nondominant HS. Open symbols represent patients who have undergone splenectomy. (Reprinted with permission from Agre P, Asimos A, Casella JF, et al. Inheritance pattern and clinical response to splenectomy as a reflection of erythrocyte spectrin deficiency in hereditary spherocytosis. N Engl J Med 1986;315:1579.)
TABLE 27.2 CLASSIFICATION OF HEREDITARY SPHEROCYTOSIS
bBy definition, patients with severe spherocytosis are transfusion-dependent.
cNormal (±SD) = 245 ± 27 × 105 spectrin dimers per erythrocyte. In most patients, ankyrin content is decreased to a comparable degree. A minority of hereditary spherocytosis patients lack band 3 or protein 4.2 and may have mild to moderate spherocytosis with normal amounts of spectrin and ankyrin.
dThe spectrin content is variable in this group of patients, presumably reflecting heterogeneity of the underlying pathophysiology.
Reprinted with permission from Walensky LD, Narla M, Lux SE. Disorders of the red blood cell membrane. In: Handin RI, Lux SE, Stossel TO, eds. Blood: principles and practice of hematology, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2003:1753.
Erythrocyte Abnormalities
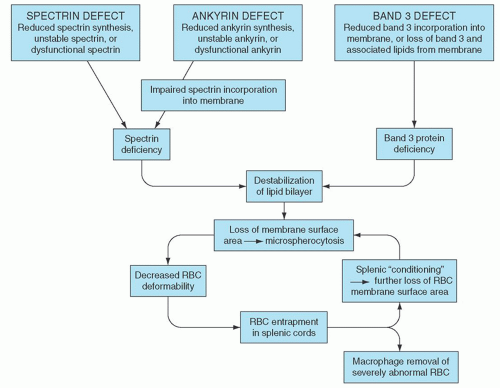
The molecular basis of HS is heterogeneous, thus it is likely that the loss of membrane surface area is a consequence of several molecular mechanisms (Fig. 27.6). The common denominator is a weakening of protein-protein interactions that link the membrane skeleton to the lipid bilayer, leading to microvesiculation, loss of membrane surface area, decreased surface/volume ratio, and spherocytosis. In cases associated with spectrin deficiency, the membrane skeleton is unable to provide adequate support for the lipid bilayer, causing destabilization of the lipid bilayer with the resultant loss of band 3-containing membrane microvesicles. In cases associated with band 3 deficiency, the lipid-stabilizing effect of band 3 is lost, releasing band 3-free microvesicles from the membrane. The loss of membrane surface area transforms red cells from biconcave discs to spherocytes with decreased cellular deformability, limiting their ability to pass through the sinusoids of the spleen and predisposing them to splenic entrapment, conditioning, and destruction.
FIGURE 27.5. Role of spectrin and ankyrin in hereditary spherocytosis. The relationship between spectrin and ankyrin content in erythrocytes of patients with dominant hereditary spherocytosis. Each point, expressed as a percentage of control (100%), represents the mean value for a kindred for both spectrin and ankyrin levels. The line represents a computer-generated fitting of the data for 19 of the 20 kindreds. The degree of spectrin and ankyrin deficiencies is essentially identical in these kindreds with one exception (open circle), an otherwise typical family in which erythrocytes are primarily ankyrin-deficient. (Reprinted with permission from Savvides P, Shalev O, John KM, et al. Combined spectrin and ankyrin deficiency is common in autosomal dominant hereditary spherocytosis. Blood 1993;82:2953.)
The membrane defect in HS leads to a variety of secondary metabolic changes. These include increased sodium and potassium flux across the HS membrane, which leads to increased membrane sodium-potassium adenosine triphosphatase activation; accelerated adenosine triphosphate (ATP) breakdown; increased glycolytic rate; decreased 2,3-diphosphoglycerate (2,3-DPG) concentration, and lower intracellular pH. HS erythrocytes are slightly dehydrated, although the reason for decreased RBC water content is not known. Possibilities include activation of K-Cl cotransport, a pathway that leads to dehydration and is activated by acid pH, possibly in the acid environment of the splenic cords, or increased Na+/K+ pump activity stimulated in response to increased passive cation leaks, leading to decreased total cation content, water efflux from the red cell, and cellular dehydration. Dehydration of HS red cells is important because it can further impair their deformability.
Role of the Spleen
The spleen plays a critical role in the pathobiology of HS, as destruction of spherocytes in the spleen is the primary cause of hemolysis in HS patients.65 The survival of infused HS red cells into normal recipients is reduced, whereas the survival of normal red cells in HS subjects is normal. Despite the persistence of spherocytosis after splenectomy, red cell survival is normal or only slightly reduced. Taken together, these observations demonstrate that an intrinsic red cell defect leads to RBC destruction, but only in the presence of an extrinsic factor—an intact spleen.
Splenic Entrapment
The unique anatomy of the splenic vasculature leads to sequestration of spherocytes selectively in the spleen. Arterial blood enters directly into the splenic cords, a network of channels formed by reticulum cells and lined by macrophages. Most of the blood that enters the splenic cords passes rapidly through direct channels, which reenter the venous system after traversing fenestrations between the lining endothelial cells. A small fraction of blood in the splenic cords percolates more slowly through this maze before reaching the venous sinuses. The hematocrit of blood from the splenic cords is high, the environment is acidic, and red cells are exposed to macrophages that line these channels. The size of fenestrations in the venous sinuses is small relative to the RBC size, and to pass through requires significant deformability of the red cell and its membrane.66 This, however, is a major problem for spherocytes, which have lost surface area and are dehydrated. Spleens removed from HS patients reveal congested cords, relatively empty venous sinuses, and few spherocytes traversing sinus walls. The most severely damaged spherocytes, cells unable to negotiate the fenestrations in the venous sinus, are removed from the circulation by macrophages. However, impaired deformability of HS red cells is only significant for the passage of these cells through the spleen. After splenectomy, red cell survival normalizes, even though spherocytes persist and sometimes are increased. Splenectomy has no effect on surface area loss by HS reticulocytes or mature red cells, because surface area loss is due to the intrinsic membrane lesion in these cells. The intrinsic membrane lesion is an independent event that continues even after splenectomy.
Splenic Conditioning
In addition to trapping HS red cells, the spleen also “conditions” these cells in a way that accelerates membrane loss and spherocyte formation. Some conditioned red cells reenter the systemic circulation, gradually shifting from osmotically normal to osmotically fragile cells during their circulation. Osmotically fragile microspherocytes are concentrated in and emanate from the splenic pulp. The more conditioned or spheroidal cells are responsible for the most fragile portion, or “the tail” of the fresh OF curve. The conditioning effect of the spleen is a cumulative injury, thought to result from several passages through the organ. The mechanism(s) of splenic conditioning is not clear. It may be the result of hemoconcentration and erythrostasis with macrophage-induced membrane injury, possibly related to the lower pH in the spleen. The calculated transit time for spherocytes is short relative to the time required for severe metabolic compromise, thus it is unlikely that metabolic depletion is important, and the ATP content of HS cells in the spleen is normal.
Clinical Features
Anemia, jaundice, and splenomegaly are the clinical features of HS most commonly encountered. However, signs and symptoms are highly variable, both with respect to the age of onset and severity.24, 25 For example, anemia or hyperbilirubinemia may be of such magnitude as to require exchange transfusion in the neonatal period or cases may escape clinical recognition altogether, presenting very late in life.67 Anemia usually is mild to moderate, but it may be absent, mild, moderate, or severe requiring transfusion. Beyond the neonatal period, jaundice is intermittent and is associated with fatigue, cold exposure, emotional distress, or pregnancy. An increase in scleral icterus and a darker urine color is commonly seen in children with nonspecific viral infections. Even when patients have no detectable jaundice, there is usually laboratory evidence of ongoing hemolysis. Splenomegaly is the rule, and in large family studies, palpable spleens have been detected in more than 75% of affected members. No apparent correlation has been shown between spleen size and disease severity, but it may exist. The liver is normal in size and function.
FIGURE 27.6. Pathophysiology of hereditary spherocytosis (HS). The primary defect in HS is a deficiency of membrane surface area, leading to the formation of spherocytes. Decreased surface area may be produced by different mechanisms: defects of spectrin and ankyrin lead to reduced density of the spectrin-actin membrane skeleton, destabilizing the overlying lipid bilayer and releasing band 3-containing microvesicles; or defects of band 3 and protein 4.2 lead to band 3 deficiency and loss of its lipid-stabilizing effect, resulting in the loss of band 3-free microvesicles. Both pathways result in loss of membrane surface area, spherocyte formation, and decreased deformability. These deformed erythrocytes become trapped in the hostile environment of the spleen cords, where splenic conditioning inflicts further membrane damage or they are removed by splenic macrophages. RBC, red blood cell.
From a clinical perspective, it has been useful to classify HS according to the severity of disease (Table 27.2).24, 25 Moderate HS is the most common presentation, recognized as a chronic hemolytic disorder with characteristic spherocytes on peripheral blood smear and an autosomal dominant pattern of inheritance.
Mild HS occurs in 20% to 30% of cases of HS. Anemia is absent, as the bone marrow is able to fully compensate for the persistent destruction of red cells, and there is little or no splenomegaly.68, 69 Because patients in this group usually are asymptomatic, they often are not diagnosed until later in life. Sometimes, they are identified as a result of hemolytic or aplastic episodes triggered by infection. Occasionally, the condition is identified through family surveys performed to document the hereditary nature of hemolytic disease in a relative.
Moderate HS accounts for 60% to 75% of all HS cases. It is associated with mild to moderate anemia, modest splenomegaly, and intermittent jaundice.68, 69 The reticulocyte count and serum bilirubin levels are elevated. Patients may require transfusion during intercurrent illness.
Severe HS occurs in ˜5% of cases of HS. It is characterized by severe hemolytic anemia, the need for red cell transfusion particularly during infancy and early childhood, and usually an incomplete response to splenectomy. The pattern of inheritance is frequently nondominant.32, 33, 35
The silent carrier of HS exists in families with autosomal recessive HS. In most cases, carriers have no signs of HS or minimal signs of HS, e.g., a slightly increased reticulocyte count, a few spherocytes on peripheral blood smear, a minimally abnormal incubated OF, or abnormal erythrocyte spectrin content detected when using sensitive techniques.32 Using a conservative estimate of 1:5,000 HS incidence in the United States, combined with the observation that ˜25% of cases are autosomal recessive, it has been calculated that the HS silent carrier state might exist in 1.4% of the U.S. population.
Hereditary Spherocytosis in Infancy
In the neonatal period, jaundice is likely to be the most prominent feature of HS. Thirty percent to 50% of adult HS patients report a history of neonatal jaundice and the diagnosis of HS is prominent in etiologic studies of severe neonatal hyperbilirubinemia.70, 71 The magnitude of hyperbilirubinemia may be severe, requiring phototherapy or exchange transfusion.72 Virtually all HS infants who are homozygous for the mutation responsible for Gilbert syndrome are jaundiced enough to require phototherapy.72 In contrast to jaundice, most newborns with HS are not anemic. Spherocytosis on the peripheral blood smear and reticulocytosis are frequently minimal or absent. However, anemia develops rapidly over the first few weeks of life in many infants and may require transfusion.72 Maturation of splenic filtering and development of the splenic circulation appear to increase the rate of hemolysis after birth; at the same time, the erythropoietic response to anemia is blunted. Within a few months, erythropoiesis increases, anemia improves, and the need for red cell transfusions disappears in all but the most severely affected infants.
Complications
The two major complications seen in HS are episodes of worsening anemia and the development of gall bladder disease.
Exacerbations of anemia occur in almost all HS patients, even in the large majority of HS patients who have mild or clinically silent disease, associated with various stresses such as infection, major surgery, trauma, pregnancy, etc. For example, previously mild anemia can become much more severe during pregnancy, usually because of the increased plasma volume, but occasionally as a consequence of accelerated hemolysis.73 As with other chronic hemolytic states, some anemic crises are preceded by a febrile illness and may be observed concurrently in more than one affected member of a single family. In some cases, there may be increased hemolysis (decreased hemoglobin, increased reticulocytes, and increased bilirubin concentration) associated with nonspecific viral infections.74
Aplastic/hypoplastic crises resulting from parvovirus B19 infection may occur, just as is seen in individuals with other chronic hemolytic disorders.75 Parvovirus B19 selectively infects erythroid precursors and inhibits their growth. Erythropoietic arrest leads to a sudden decrease in hemoglobin concentration and reticulocytopenia. Recovery occurs within 7 to 10 days and is heralded by reticulocytosis and thrombocytosis. Parvovirus infection may be the first manifestation of HS, and multiple HS family members infected with parvovirus have developed aplastic crises at the same time, leading to descriptions of “epidemics” of HS.76 Infection with parvovirus is a particular threat to susceptible pregnant women, because it can infect the fetus, leading to severe anemia, hydrops fetalis, and fetal death.
Less commonly, exaggeration of anemia is the result of exhaustion of folate reserves by the sustained increase in net DNA synthesis.77 Megaloblastic arrest of erythropoiesis has been observed most commonly during pregnancy, in association with liver disease, or in patients recovering from an aplastic crisis.
Cholelithiasis is common in HS just as in other chronic hemolytic disorders. Bilirubin pigment gallstones may be found in infants and young children,78 but the incidence of gallstones increases markedly with age, and they are present in 40% to 80% of adults. The history of family members with cholelithiasis in the second or third decade of life is a clue to the possibility of HS or another inherited hemolytic disorder. In patients with mild HS, cholelithiasis may be the initial presentation of the disease. The development of gallstones is increased approximately fivefold in HS patients who are homozygotes for the uridine diphosphateglucurono-syltransferase mutation responsible for Gilbert syndrome.79, 80 Because of their high incidence, HS patients should be periodically examined by ultrasound for the presence of gallstones, beginning early in childhood.
A few other unusual complications have been noted in HS patients. Heterotopia of the bone marrow has been noted rarely in the renal pelvis or along the vertebral column.81 These extramedullary masses of marrow may be mistaken for malignant tumors. After the spleen is removed, they undergo fatty metamorphosis. Hemosiderosis and multiple endocrine disorders resulting from transfusion-induced iron overload have been described. Interestingly, symptomatic iron overload also has been reported in some untransfused HS patients who are heterozygous for the hemochromatosis gene. Cases of HS and hematologic malignancy, including multiple myeloma, leukemia, and myeloproliferative disorders, have been reported, suggested to be linked to the persistent hematopoietic stress of HS; but the relationship with HS, if more than coincidental, remains to be determined. Gout and chronic leg ulcers/dermatitis are unusual complications in adults with HS.82, 83 Other rare complications include thrombosis, spinocerebellar degenerative syndromes, movement disorder with myopathy, and hypertrophic cardiomyopathy.84
Laboratory Features
The classic laboratory features of HS include anemia, reticulocytosis, increased mean corpuscular hemoglobin concentration (MCHC), spherocytes on the peripheral blood smear, hyperbilirubinemia, and an abnormal OF test.
Anemia is typically mild, accompanied by reticulocytosis with values of 5% to 20%. This compensation is associated with increased erythropoiesis and elevated levels of erythropoietin.68, 69 Erythrocyte indices demonstrate a normal or borderline low mean corpuscular volume (MCV) despite increased numbers of reticulocytes, reflecting the membrane loss and dehydration. The MCHC is usually increased (≥35%) due to mild cellular dehydration.85, 86 Examination of indices obtained by automated cell counters can be used as a screening test for HS. In unsplenectomized children, an MCHC >35.4/dl and a red cell distribution (RDW) width >14 had a sensitivity of 63% and a specificity of 100%86 for the diagnosis of HS. Histograms of hyperdense erythrocytes (MCHC > 40 g/dl) obtained from laser-based cell counters have been used as a screening test for HS, and when combined with an elevated MCHC, have been claimed to identify nearly all cases of HS.
Only gold members can continue reading. Log In or Register to continue
Oct 21, 2016 | Posted by drzezo in HEMATOLOGY | Comments Off on Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane