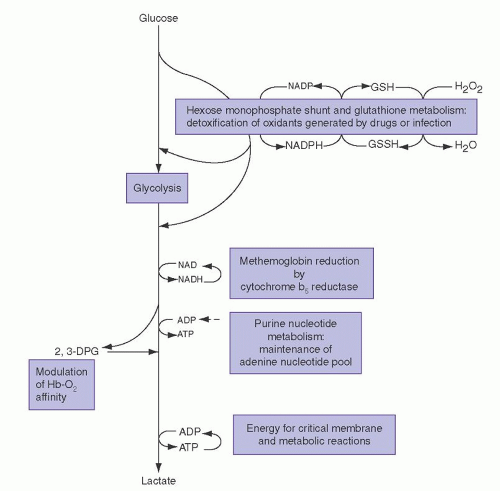
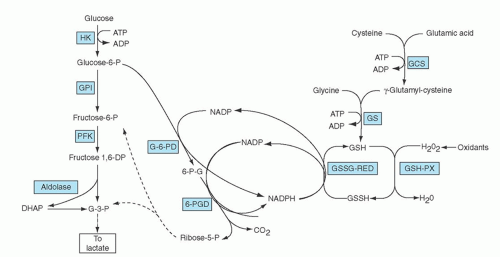
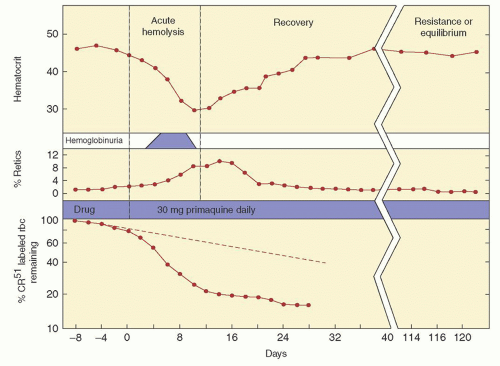
The importance of this enzyme for red cell integrity was first recognized following the observation that some African-American soldiers taking the antimalarial drug primaquine would develop acute hemolytic anemia with hemoglobinuria. Initially it was observed that GSH was decreased in the RBC of susceptible individuals during acute hemolytic episodes. Subsequently, the activity of G6PD, one of the enzymes needed to keep adequate GSH levels, was found to be deficient in affected red cells.
5,
6 Soon thereafter, the worldwide distribution of G6PD deficiency became apparent, and the variation of clinical expression of enzyme deficiency was discovered. In most individuals with G6PD deficiency, there is no anemia in the steady state, reticulocyte counts are normal, but RBC survival may be slightly decreased. However, episodic exacerbations of hemolysis accompanied by anemia occur in association with the administration of certain drugs, with some infections, and with the eating of fava beans. In a minority of cases G6PD deficiency is associated with a chronic hemolytic process. To date, over 400 G6PD biochemical variants have been recognized.
2,
3,
7
Genetics
The gene for G6PD is located on the X chromosome (band X q28),
8,
9,
10 The fact that normal males and females have the same enzyme activity in their red cells is explained by the Lyon hypothesis.
1,
11 This hypothesis maintains that one of two X chromosomes in each cell of the female embryo is inactivated and remains inactive throughout subsequent cell divisions for the duration of life. In fact, it was studies by Beutler on females with G6PD deficiency which were used in proof of the Lyon hypothesis.
12 Enzyme deficiency is expressed in males carrying a variant gene, whereas heterozygous females usually are clinically normal. However, dependent upon the degree of lyonization, and the degree to which the abnormal G6PD variant is expressed, the mean red blood cell enzyme activity in females may be normal, moderately
reduced, or grossly deficient. A female with 50% normal G6PD activity has 50% normal red cells and 50% G6PD-deficient red cells. The G6PD-deficient cells in females, however, are as vulnerable to hemolysis as are enzyme-deficient red blood cells in males.
The concept of X chromosome inactivation and the study of G6PD has been informative in other areas. In particular, it has facilitated our understanding of monoclonal and multiclonal disorders of cell proliferation. Most tumors, both benign and malignant, can be demonstrated to have a clonal origin, being derived from one cell.
13,
14,
15 For example, analysis of the G6PD enzyme in uterine myomata of women heterozygous for G6PD A and G6PD B revealed that any given tumor had either G6PD A or enzyme B, but not both.
16 The same principle has been used to demonstrate the clonal origin of the malignant transformation of acute leukemia
17,
18 and chronic myelocytic leukemia,
19 and also the clonal nature of polycythemia vera,
20 primary thrombocythemia,
21 and paroxysmal nocturnal hemoglobinuria.
22
Prevalence and Geographic Distribution
Deficiency of G6PD is the most common metabolic disorder of red blood cells and has been estimated to affect over 400 million people worldwide.
2,
3,
23 Although global in its distribution, G6PD deficiency is encountered with greatest frequency in the tropical and subtropical zones of the Eastern Hemisphere. The incidence of the deficiency state is approximately 20% among African Bantu males,
24,
25 12% in African-American men,
26 and 8% in Brazilian blacks. As many as 20% of female African-Americans may be heterozygous for G6PD mutants,
27 and as many as 1% are homozygous. In Sardinia, the incidence varies from 35% at low altitudes to 3% in areas above 600 meters.
28 The deficiency state has been reported from most areas of Greece, again with greatest frequency (20% to 32%) in the lowlands.
29 The condition is also prevalent among Sephardic Jews, and as many as 60% to 70% of Kurdish Jews may be affected.
3,
30,
31 In the male Asian population, the incidence of G6PD deficiency is estimated to be 14% in Cambodia,
32 5.5% in South China,
33,
34 2.6% in India,
32 and less than 0.1% in Japan.
35 In China the frequency of G6PD deficiency in males ranges from 0% to 17.4%, with the highest prevalence being seen in ethnic groups geographically related to historical malaria. It is rare among Native Americans.
36Because of its high incidence among populations in which malaria was once endemic, G6PD deficiency is thought to have conferred a selective advantage against infection by falciparum malaria.
37,
38,
39 Partial indirect support for this is that G6PD deficiency in Sardinia is more common at sea level compared to higher elevations, and this also parallels the endemicity of malaria. In addition, it has been observed that parasitized female heterozygotes for G6PD deficiency (who therefore have normal and G6PD-deficient RBC) have more malaria parasites in normal erythrocytes compared to their own G6PD-deficient cells.
40 Moreover, it has been demonstrated that the in vitro growth of malarial parasites is inhibited in G6PD-deficient red cells.
41 The
precise reasons for the observed inhibition of parasite growth in G6PD-deficient red cells are not known. One possibility is that the oxidant stress which causes GSH instability, and destroys the host RBC, also kills the parasite.
2,
42,
43,
44
The Enzyme and Its Variants
The monomeric form of G6PD contains 515 amino acids and has a molecular weight of 59 KDa.
45,
46 The active form of G6PD in vivo is a dimer which requires NADP for its stability.
47 The G6PD gene has been cloned and sequenced. It is known to contain 13 exons, and is over 18 kb in length.
46,
48,
49The normal or wild-type enzyme is G6PD B, although hundreds of variant enzymes now have been identified. By international agreement, standardized methods have been used to characterize these enzyme variants, which differ on the basis of biochemical properties such as kinetic activity, electrophoretic mobility, the Michaelis constant for its substrate glucose-6-phosphate and cofactor NADP, the ability to utilize different substrate analogues, heat stability, and pH optima. Throughout the years the published list of recognized G6PD biochemical variants has been periodically updated,
50,
51 and over 400 biochemical variant forms of G6PD are recognized.
3 However, differences between some variants are subtle, and most likely reflect minor technical differences between laboratories rather than true enzyme differences. Moreover, advances in molecular biology have revealed that many biochemical G6PD variants, in fact, have the same DNA defect (see below).
The World Health Organization (WHO) has classified G6PD variants on the magnitude of the enzyme deficiency and also the severity of hemolysis.
52 Class I variants have very severe enzyme deficiency (less than 10% to.20% of normal) and have chronic hemolytic anemia.
Class II variants also have severe enzyme deficiency (less than 10% of normal), but there is usually only intermittent hemolysis.
Class III variants have moderate enzyme deficiency (10% to 60% of normal) with intermittent hemolysis usually associated with infection or drugs.
Class IV variants have no enzyme deficiency or hemolysis.
Class V variants are those in which enzyme activity is increased. Variants in the last two groups, although of much interest to biologists, geneticists, and anthropologists, are of no major clinical significance.
The normal wild-type enzyme, G6PD B, is found in most Caucasians, Asians, and a majority of blacks. It has normal catalytic activity and is not associated with hemolysis (Class IV). A commonly encountered variant is G6PD A
+ which is found in 20% to 30% of blacks from Africa.
53 It has normal catalytic properties and does not cause hemolysis (Class IV). It differs from G6PD B in that it has a much faster electrophoretic mobility (the letters A and B refer to relative electrophoretic mobilities). The structure of G6PD A
+ differs from that of G6PD B by the substitution of a single amino acid, an asparagine for aspartate at the 126th position of the protein.
54 Another common variant, G6PD A
–, is the enzyme responsible for primaquine sensitivity in blacks, and it is the most common variant associated with mild to moderate hemolysis (Class III). This G6PD variant is found in 10% to 15% of African-Americans, and with similar frequencies in western and central Africa.
55 It has an electrophoretic mobility identical to that of G6PD A
+. However, this is an unstable enzyme and its catalytic activity, although nearly normal in bone marrow cells and reticulocytes,
56 decreases markedly in older RBC.
57 Hence, this variant is designated G6PD A
– compared with G6PD A
+ (the + and – denote enzyme activity). G6PD Mediterranean is a common abnormal variant found in people whose origins are in the Mediterranean area. However, this same variant also is found in the Middle East and India.
2 The electrophoretic mobility of G6PD Mediterranean is identical to that of G6PD B, but its catalytic activity is markedly reduced, and hemolysis can be severe (Class II).
56 G6PD Canton is a common variant enzyme seen in Asians.
58 Its biochemical properties are very similar to those of G6PD Mediterranean.
Advances in molecular biology have further enhanced our understanding of G6PD deficiency, and now over 160 different gene mutations or mutation combinations have been identified.
2,
59,
60,
61 These DNA changes almost all are missense mutations leading to single amino acid substitutions in the enzyme. Large deletions have not been identified, suggesting that complete absence of G6PD might be lethal.
3 The mutations are located throughout the entire coding region of the gene.
62 However, in Class I variants associated with chronic hemolysis, mutations are clustered around exon 10, an area that governs the formation of the active G6PD dimer.
62,
63 The correlation between the different biochemical variants, the site of genetic mutation, and the extent of hemolysis is a matter of current investigation.
2An interesting example of how molecular biology has enhanced our understanding relates to G6PD A
–, once thought to be a single unstable variant found in blacks throughout the world. However, molecular analysis now has demonstrated that G6PD A
– may have more than one genotype. In all cases there is a mutation at nucleotide 376 (A→G), which also is the nucleotide substitution characteristic of G6PD A
+. In addition, the G6PD A
– variants have a second mutation, and in the majority of cases it is at nucleotide 202 (G→A).
3,
64 A smaller fraction of G6PD A
– subjects have the second substitution at nucleotide 680 (G→T) or at nucleotide 968 (T→C).
65 Thus, the G6PD A
– variant, once thought to be a single homogeneous mutation in Africans, now turns out to represent at least three different genotypes.
3 Also, a number of G6PD variants originally described in non-Africans are now found to have one of the known G6PD A
– mutations (
Table 28.1). For example, G6PD Betica,
66 a Spanish variant and G6PD Matera,
67 an Italian variant, have demonstrated base substitutions at nucleotides 202 and 376, identical to the common G6PD A
– variant. They are examples, therefore, of G6PD A
–. One subject with G6PD Betica had base substitutions at nucleotides 376 and 968, identical to the less common G6PD A
– variant.
66There are several other variants that appear clinically and biochemically heterogeneous but have been found to be genetically more uniform. For example, G6PD Mediterranean involves many different ethnic groups, although most subjects have the same genetic defect, a single base substitution (C→T) at nucleotide 563.
66,
67,
68 Moreover, just as in the case of G6PD A
–, many of the different biochemical variants have turned out to have the same molecular defect as G6PD Mediterranean (
Table 28.1).
In China there are at least 21 variants causing G6PD deficiency. The three most common are G6PD Canton (G→T at nucleotide 1,376), G6PD Kaiping (G→A at nucleotide 1,388), and G6PD Gaohe (A→G at nucleotide 95).
69G6PD Canton and G6PD Gaohe are mainly regarded as WHO Class II variants, while G6PD Kaiping is considered a WHO Class III variant.
Because leukocyte and platelet G6PD is regulated by the same gene as that of red cells, documentation of decreased activity in the white blood cells
70,
71,
72 and platelets
71 of deficient individuals is not surprising. Because of the normally short survival of leukocytes and platelets, however, most individuals with G6PD deficiency do not manifest impairment of phagocytosis or bactericidal activity of granulocytes.
71,
72 The exception to this occurs with Class I G6PD deficiency, where some affected individuals may have neutrophil dysfunction and increased susceptibility to infection.
73,
74
Screening
Aside from the District of Columbia, there is no generalized neonatal screening program for G6PD deficiency in the United States. The approach here has been to recognize that neonatal hyperbilirubinemia may be caused by G6PD deficiency and these children should be tested and monitored closely. In some countries where the predominant G6PD variants are Class II mutations, screening programs have been instituted. Neonatal screening for G6PD deficiency has been very effective in reducing the incidence of favism later in life in Sardinia
145 and other regions where this potentially fatal complication is common.
112 Prenatal diagnosis utilizing molecular techniques is potentially available, but the benign course of most G6PD variants has precluded its development.
146Routine blood bank screening has been considered unwarranted and G6PD deficiency is not considered a problem in transfusion medicine. Even in areas where G6PD deficiency is endemic, screening of blood donors is not required. One careful evaluation of the recipients of G6PD-deficient blood uncovered no deleterious consequences.
147 However, patients receiving G6PD Mediterranean blood may have an increased serum bilirubin and lactate dehydrogenase (LDH) concentration following transfusion, and this can be confused with a transfusion reaction.
148 In premature infants, simple transfusions with G6PD-deficient red cells have been associated with hemolysis and severe hyperbilirubinemia requiring exchange transfusion.
149 Also, massive intravascular hemolysis has occurred in an Indian neonate following an exchange transfusion with G6PD-deficient blood.
150 In view of these occurrences, it has been recommended that in areas where G6PD deficiency (presumably Class II variants) is common, donor blood should be screened for the enzyme before transfusing premature infants
149 or using the blood for a neonatal exchange transfusion.
150 This recommendation currently is not standard blood banking practice.