Hemolytic disease of the fetus and newborn (HDFN) results from the destruction of red blood cells by maternal immunoglobulin (Ig) G antibodies that gain access to the fetal circulation during pregnancy. These antibodies may be directed against Rhesus (Rh) or other blood group antigens on fetal red blood cells, inherited from the father and not present on the mother’s red blood cells. HDFN associated with blood group antibodies has a broad spectrum of effects, ranging from mild anemia and hyperbilirubinemia in an infant to life-threatening complications before birth.1 Clinical manifestations of severe hemolytic disease before birth include profound fetal anemia, hepatosplenomegaly, generalized edema, massive ascites, and congestive heart failure. The accelerated destruction of fetal red blood cells elicits extramedullary hematopoiesis and release of nucleated and other immature red blood cells into the peripheral circulation (erythroblastosis fetalis). Ongoing hemolysis of red blood cells after birth results in neonatal hyperbilirubinemia that may cause kernicterus and permanent neurologic injury or death.
HISTORICAL BACKGROUND
In the 20th century, HDFN was recognized as a distinct clinical entity, its pathogenesis was delineated, and an effective strategy for preventing its most common form was introduced. Case reports in the early 1900s (and before) described edematous (hydropic) stillborns and anemic infants with marked jaundice who died within days of birth. Not until 1932 were these phenomena realized to be the same hematologic disease process when Diamond et al. described the interrelationship of neonatal anemia, jaundice, and edema as symptoms that occur in varying degrees and combinations in erythroblastosis fetalis.2
The precise etiology of the disease remained obscure until seminal observations on blood group antigens and incompatibility in pregnancy were made in the 1940s. Landsteiner and Weiner first used immune sera raised in rabbits against red blood cells from Rhesus monkeys to agglutinate human red blood cells, thus discovering the Rhesus factor.3 This antigen, now known as the D antigen of the Rhesus blood group in humans, is present exclusively on red blood cells of Rh-positive individuals. In the year before this discovery, Levine and Stetson recognized that a woman could become immunized against paternally inherited red blood cell determinants of the fetus during pregnancy.4 Using Landsteiner’s anti-Rh antisera to investigate cases of erythroblastosis fetalis, Levine et al. subsequently demonstrated that 90% of the mothers were Rh negative, and all the fathers and infants were Rh positive.5 This statistical association as well as the presence of Rh agglutinins in the blood of mothers with affected infants supported their theory that alloimmunization and transplacental passage of these antibodies caused destruction of fetal red blood cells.5 The inciting stimulus for red blood cell alloimmunization in pregnancy, the passage of fetal red blood cells into the maternal circulation or fetal-maternal hemorrhage (FMH), was directly demonstrated by Chown in 1954.6
Methods to monitor alloimmunized pregnancies and to treat affected fetuses and infants were first introduced in the late 1940s. Neonatal exchange transfusion enabled simultaneous correction of the anemia and reduction of bilirubin concentration in affected infants.1, 7 In 1961, Liley described the relationship between the concentration of bilirubin in amniotic fluid and the degree of destruction of fetal red blood cells, providing a tool to assess the severity of intrauterine hemolytic disease.8 Two years later, Liley introduced the technique of intraperitoneal transfusion of anemic fetuses, which was used for more than 20 years before it was largely supplanted with intravascular transfusion techniques.9
Primary prevention of D alloimmunization became possible with the advent of anti-D immune prophylaxis. The ability of passively transferred antibodies to effectively block active immunization to foreign antigens was first demonstrated by Von Dungern in 1900.1 Experimental studies in the 1960s applied this approach to D alloimmunization, revealing that Rh-negative men could be protected if administered anti-D immune globulin (RhIG) before transfusion with Rh-positive red blood cells.10, 11 Between 1963 and 1968, clinical trials involving Rh-negative pregnant women demonstrated that administration of RhIG within 72 hours of delivery was successful in reducing the incidence of D alloimmunization from 7% to 15% to 1% to 2%.12, 13, 14, 15, 16, 17 Metaanalysis of clinical trials of postpartum RhIG administration confirms a >90% reduction in the alloimmunization rate among treated women compared to untreated women (Table 30.1).17 The recognition that FMH occurring primarily in the third trimester contributed to residual risk of alloimmunization during pregnancy led to the clinical observation that additional, antenatal RhIG prophylaxis could further reduce the risk of D alloimmunization to <1%.18, 19 Meta-analysis of two randomized controlled trials involving more than 4,500 women confirms the effectiveness of antenatal administration of RhIG at 28 weeks and 32 weeks of pregnancy (Table 30.1).20, 21, 22
By 1971, the administration of RhIG to D-negative women after delivery of a D-positive infant and after abortion was recommended by the World Health Organization and rapidly became widespread practice.23 Recommendations for antenatal administration of RhIG were more controversial in some countries because of supply concerns and economic cost-benefit arguments, but were introduced in Canada and the United States in 1979 to 1980 and in the United Kingdom in 1998.24, 25, 26, 27 In the United States, the incidence of HDFN due to anti-D decreased from 40.5 to 10.6 cases per 10,000 total births between 1970 and 1986.28, 29 RhIG accounted for most of this improvement, but the trend toward smaller families and improved quality of perinatal care also contributed to the decline in the incidence of anti-D in pregnant women and related perinatal mortality, respectively.30
Despite all preventive efforts, HDFN due to anti-D continues to occur in about 6.7 of 1,000 live births in the United States.31 Failure to prevent maternal D alloimmunization is usually due to inadvertent failure to administer RhIG, or, less commonly, to production of anti-D antibodies early in pregnancy before an antenatal dose of RhIG. No prophylactic measures are available to prevent sensitization to other blood group antigens in pregnancy, most notably Rhesus (c), Kell (K1), and Duffy (Fya), and the corresponding maternal alloantibodies can cause severe HDFN.32, 33
PATHOPHYSIOLOGY
The risk of maternal alloimmunization to blood group antigens is related to the frequency of blood group alleles in the population, volume of incompatible red blood cell exposure, immunogenicity of the sensitizing red blood cell antigen, and maternal immune responsiveness. The propensity of maternal red blood cell antibodies to cause HDFN and the severity of the disease in individual cases are affected by the inherent characteristics of the red blood cell antibodies and the compensatory physiologic reaction to the anemia in the infant.
TABLE 30.1 META-ANALYSIS OF ANTI-D IMMUNE GLOBULIN (RHIG) CLINICAL TRIALS
Treatment
Control
Odds Ratio, 95% Confidence Interval
Postpartum RhIG (Treatment) vs. No Treatment (Control)
Immunization after 6 mo (5 RCTs)
10/4,756 (0.2%)
204/2,824 (7%)
0.08 (0.06, 0.11)
Immunization in a subsequent
11/682 (1.6%)
57/389 (14.6%)
0.12 (0.70, 0.19)
pregnancy (4 RCTs)
Postpartum and Antenatal RhIG (Treatment) vs. Postpartum RhIG (Control)
Immunization in pregnancy after birth of Rh-positive infant (2 RCTs)
5/1,112 (0.4%)
13/1,185 (1.1%)
0.44 (0.18, 1.12)
Immunization at 2-12 mo, primigravidae (1 RCT)
0/362 (0)
4/360 (0.7%)
0.13 (0.02, 0.96)
RCT, randomized controlled trial.
Data from Lee D, Rawlinson VI. Multicentre trial of antepartum low dose anti-D immunoglobulin. Transfus Med 1995;5:15-19; and Hensleigh PA. Preventing rhesus isoimmunization: antepartum Rh immune globulin prophylaxis versus a sensitive test for risk identification. Am J Obstet Gynecol 1983;146:749-755; Crowther C, Middleton P. Anti-D administration after childbirth for preventing Rhesus alloimmunization. Cochrane Database Syst Rev. 2000;(2):CD000021.
Fetal-Maternal Hemorrhage
Although the fetal circulation is separated from the maternal circulation by placental membranes and fetal capillaries, blood cells pass between the fetal and maternal bloodstreams throughout gestation. FMH occurs in 3% of pregnancies in the first trimester, 12% in the second trimester, 45% in the third trimester, and 64% to 100% after delivery.34, 35 The total volume of fetal cells in the maternal circulation is usually small and does not exceed 0.1 to 0.25 ml in most cases.36 Large-volume FMH occurs less often, with more than 15 ml of fetal red blood cells (approximately 30 ml whole blood) detected at a rate of 1.6% after cesarean section or complicated vaginal delivery and 0.7% after spontaneous vaginal delivery.37 Invasive procedures, clinical maneuvers, or other traumatic events during pregnancy may also elicit sufficient FMH to induce or augment the production of alloantibodies against red blood cells in susceptible pregnant women (Table 30.2).38, 39, 40
Maternal Alloimmunization to Blood Group Antigens
The likelihood of a relevant blood group incompatibility occurring in pregnancy depends on the frequency of blood group alleles in the population.41 Among Caucasians and African Americans in the United States, approximately 15% and 8%, respectively, of women are D negative and lack a functional D gene on both chromosomes.41 Incompatibility with respect to the D antigen occurs in approximately 10% of pregnancies; among D-negative women, approximately 60% to 70% of pregnancies yield D-positive infants. American Indians and Asians are almost all D positive; consequently, D alloimmunization is extremely rare among these populations.41 Although virtually all pregnant women are exposed to fetal red blood cells with childbirth, alloimmunization to blood group antigens occurs in only a fraction of incompatible pregnancies. Approximately one in six multiparous D-negative women developed anti-D antibodies without RhIG prophylaxis in incompatible pregnancies. The risk of alloimmunization to the D antigen has been estimated for other obstetric interventions (Table 30.2).38, 39, 40
TABLE 30.2 FETAL-MATERNAL HEMORRHAGE AND RHD ALLOIMMUNIZATION: RATES AMONG RH-NEGATIVE WOMEN WITH D-POSITIVE INFANTS, WHO DO NOT RECEIVE ANTI-D IMMUNE GLOBULIN
Fetal-Maternal Hemorrhage (%)
Primary RhD Alloimmunization (%)
Pregnancy and delivery
Before 29 week of gestation
3-12
0.3-1.9
35 week to delivery
65-100
7-15
Abortion
Induced abortion
4-30
4-5
Spontaneous abortion
3-12
1.5-2.0
Threatened abortion
10
ND
Ectopic pregnancy
24
Case reports
Amniocentesis
7-15
2-5
Cordocentesis
57
ND
Chorionic villus sampling
14
1-2
External cephalic version
2-28
ND
Abdominal trauma
28
Case reports
ND, not determined.
Data from Huchet J, Dallemagne S, Huchet C, et al. The antepartum use of anti-D immunoglobulin in rhesus negative women. Parallel evaluation of fetal blood cells passing through the placenta. The results of a multicenter study carried out in the region of Paris. J Gynecol Obstet Biol Reprod (Paris) 1987;16:101-111; Medearis AL, Hensleigh PA, Parks DR, et al. Detection of fetal erythrocytes in maternal blood postpartum with the fluorescence-activated cell sorter. Am J Obstet Gynecol 1984;148:290-295; Ness PM, Baldwin ML, Niebyl JR. Clinical high-risk designation does not predict excess fetal-maternal hemorrhage. Am J Obstet Gynecol 1987;156:154-158; American College of Obstetricians and Gynecologists. Prevention of RhD alloimmunization. Washington, DC: ACOG Practice Bulletin No. 4, May 1999.
A primary determinant of the risk of red blood cell alloimmunization is the volume of incompatible red blood cell exposure. Less than 1 ml of D-positive, fetal red blood cells is sufficient to induce anti-D antibody formation in 0.3% to 1.9% of D-negative women before delivery, whereas transfusion of a unit of D-positive red blood cells (300 ml) immunizes approximately 70% of D-negative individuals.16, 41 Host factors also influence the risk of red blood cell alloimmunization. Concomitant ABO and D incompatibility between the mother and fetus (e.g., a type O, D-negative mother with a type A, B, or AB, D-positive fetus) results in an almost ninefold reduction in the risk of alloimmunization to the D antigen in a first pregnancy.18, 42 ABO incompatibility does not prevent a secondary immune response in a sensitized individual in subsequent pregnancies. The D antigen is one of the most potent immunogens among red blood cell antigens, but even after incompatible blood transfusion or multiple D-positive pregnancies, approximately 30% of D-negative individuals do not produce anti-D antibodies and are called “nonresponders.”41 Complex genetic factors regulate immune responses, and the basis of this variability to red blood cell immunization among individuals is not well understood.
The overall frequency of alloimmunization to clinically significant blood group antigens among women ranges from 0.04% to 0.3%.43, 44, 45, 46, 47 The variability in these rates may be due to geographic differences in blood group antigen expression, national blood transfusion practice, and higher-order birth and abortion rates in the population, as well as the sensitivity of laboratory methods used in prenatal antibody screening.43, 44, 45, 46, 47, 48 Anti-D is still among the most frequently detected antibodies in sensitized pregnancies despite a precipitous decline in its incidence after the introduction of RhIG prophylaxis (Table 30.3).43, 44 In recent decades, other red blood cell alloantibodies have accounted for proportionately more cases of maternal alloimmunization and HDFN. The K1 antigen surpassed the D antigen as the leading cause of alloimmunization among women in a recent series, occurring at a rate of 3.2 in 1,000 compared to 2.7 in 1,000 for anti-D.43 Women who develop anti-K1 antibodies in pregnancy often have a history of blood transfusion as the immunizing stimulus.48 In another series of 1,133 Dutch women with positive antibody screens, anti-E was the most common antibody detected (23%) followed by anti-K (18.8%), anti-D (18.7%), and anti-c (10.4%).47
ABO Blood Group Isoagglutinins
ABO incompatibility occurs statistically in one of every five pregnancies. ABO isoagglutinins are present in the sera of all individuals whose red blood cells lack the corresponding antigen and are usually of the IgM class. High titers of IgG antibodies are more likely to occur in group O individuals than in group A or B individuals, and increased antibody production after antigenic stimulation can occur.41 Consequently, group O mothers with potent IgG anti-A, -B, or -A,B are at greatest risk of having affected infants. Among group A or B infants born to group O mothers, 30% to 50% have detectable maternal IgG antibody bound to their red blood cells compared to 5% among all infants.49 Because ABO IgG antibodies can occur without prior red blood cell exposure, they can result in hemolytic disease of the newborn (HDN) in a first pregnancy. However, ABO antigens are not fully developed on red blood cells at birth, and similar carbohydrate antigens occur on other tissues that effectively neutralize anti-A, anti-B, and anti-A,B antibodies to a large extent, thereby mitigating their effect. At birth, neonatal anemia due to ABO HDN is usually mild. Antibodies directed against other carbohydrate blood group antigens (Lewis, I, P) are naturally occurring IgM antibodies that are inconsequential in pregnancy because IgM is not transported across the placenta.
TABLE 30.3 RED BLOOD CELL ALLOANTIBODIES: OCCURRENCE IN WOMEN AND ASSOCIATION WITH HEMOLYTIC DISEASE OF THE FETUS AND NEWBORN (HDFN)
Other Rh specificities in rare cases of moderate to severe fetal anemia: Cw, Cx, Goa, RH36 (Berrens, Bea), RH37 (Evans), RH32
Kell
K1
2.1
3.0
Mild to severe HDFN, hydrops fetalis
K2
0
0.03
Other Kell specificities in rare cases of moderate to severe fetal anemia: k ([Cellano; K2] [rare]), Jsa, Jsb, Ku
Lewis (Lea, Leb)
2.2
3.0
Not a cause of HDFN
Duffy
Fya
0.4
0.8
Mild to severe HDFN, hydrops fetalis
Fyb
0.02
0.03
Not a cause of HDF; no or mild HDN
MNS
M
0.7
0.5
Rare cases of moderate to severe HDF
N
0.1
0.03
Not a cause of HDF; no or mild HDN
S
0.2
0.1
Rare cases of moderate to severe HDF
Other
Other MNS specificities in rare cases of moderate to severe fetal anemia: s, U, Mta
Lutheran (Lua, Lub)
0.02
0.1
No or mild HDFN
Ii
0.3
0.1
Not a cause of HDFN
Kidd
Jka
0.2
0.2
Mild to severe HDFN
Jkb
0
0
Mild to severe HDFN (rare)
P (P1)
0.6
0.03
Not a cause of HDFN
Other P group specificities in rare cases of moderate to severe fetal anemia: PP1Pk
Other
0.3
0.03
HLA (Bga, Bgb, Bgc): Not a cause of HDFN
Other blood group specificities in rare cases of moderate to severe fetal anemia: Diego (Dia), Cartwright (Yta), Biles (Bi), Radin (Rd), Wright (Wra)
Fetal and Neonatal Immune-mediated Anemia
In contrast to naturally occurring antibodies against carbohydrate blood group antigens, antibodies against Rhesus, Kell, and other protein blood group antigens only rarely occur without exposure to incompatible red blood cells.41 IgM may be detected in the primary immune response after the initial stimulus, but IgG may be the only immunoglobulin found and is typically present 5 to 15 weeks after FMH. Because FMH usually occurs late in the third trimester or at delivery, IgG antibodies against red blood cell alloantigens usually do not reach appreciable concentration to cause significant disease in a first pregnancy. Upon re-exposure to the red blood cell antigen, however, an anamnestic immune response rapidly produces IgG with enhanced avidity for target fetal red blood cells, translating to earlier onset and greater severity of hemolytic disease in subsequent incompatible pregnancies.
Maternal IgG red blood cell antibodies bind to their target red blood cells and cause Fc-receptor-mediated extravascular destruction by splenic macrophages and other cytotoxic effector cells in the reticuloendothelial system. Progressive removal of portions of the red blood cell membrane by macrophages and other phagocytic cells in the spleen can produce spherocytes in the peripheral circulation. Anti-D is usually high-affinity IgG1 and IgG3, which are the typical IgG subclass responses to protein antigens, and involve T-helper (Th) cells for their production.50 The immunoglobulin subclasses IgG1 and IgG3 have greater affinity for Fc receptors on phagocytic cells than IgG2 and IgG4, which may account for their association with more severe hemolytic disease.41, 50 The concomitant presence of maternal antihumanleukocyte antigen (HLA) and anti-D is associated with a mild course of HDFN, possibly due to the competitive binding of the HLA antibodies to Fc receptors on cytotoxic effector cells.51
Extravascular destruction of red blood cells often cannot account completely for the degree of fetal anemia due to anti-K1, unlike most cases of HDF due to anti-D and other red blood cell alloantibodies. Severe fetal anemia may occur with anti-K1 despite low maternal antibody titers and falsely reassuring concentrations of bilirubin in amniotic fluid.48, 52 In Kell-sensitized pregnancies, affected fetuses often have fewer reticulocytes and normoblasts in their peripheral circulation than infants with comparable anemia caused by anti-D, which suggests impaired production of red blood cells.53, 54 Anti-K1 was shown to inhibit specifically the growth of K1-positive erythroid progenitor cells in vitro, whereas anti-D exhibited no effect with D-positive precursors in the same assay.55 These observations support the dual action of maternal anti-Kell in eliciting both peripheral red blood cell destruction and erythropoietic suppression to produce fetal anemia.
Most cases of severe fetal anemia are caused by anti-D, anti-c, and anti-K1; rarely, anti-Fya or other IgG red blood cell alloantibodies are associated with severe HDFN (Table 30.3). Compensatory hematopoiesis in the bone marrow and extramedullary hematopoiesis primarily in the liver and spleen result in the release of nucleated red blood cells, reticulocytes, normoblasts, and other immature erythrocytes in the fetal circulation. Severely affected fetuses have marked hepatosplenomegaly due to the extramedullary hematopoiesis, which can lead to portal and umbilical venous obstruction, portal hypertension, and hepatocellular damage.1 Production of albumin and other plasma proteins by the liver is markedly impaired, and hypoproteinemia results. Severe anemia and hepatic dysfunction with hypoproteinemia and portal hypertension may also lead to the development of congestive heart failure. Hydrops fetalis describes the ultimate outcome of these physiologic insults, with the development of generalized edema (anasarca), marked ascites, and pleural and pericardial effusions. Although the pathogenesis of hydrops fetalis is not clearly defined, the extent of hepatic damage rather than the degree of anemia more consistently correlates with the severity of the condition.1
Neonatal Immune-mediated Hyperbilirubinemia
Immune-mediated destruction of fetal red blood cells results in increased serum concentration of free heme, which is further metabolized to unconjugated (indirect) bilirubin. During gestation, unconjugated bilirubin and other metabolites are transported across the placenta and eliminated by the mother. When the umbilical cord is severed at birth, unconjugated bilirubin begins to accumulate because infants have immature liver function and are not capable of efficiently metabolizing bilirubin.
Unconjugated bilirubin is transported in the plasma bound to albumin, but when its concentration exceeds the plasmabinding capacity or when it is displaced from carrier proteins, the lipophilic free molecule can cross cell membranes and impair mitochondrial function to cause cell death. Preterm infants are at greater risk for developing bilirubin encephalopathy than are term infants because of the immaturity of their blood-brain barrier as well as their more pronounced hepatic deficiency. Bilirubin toxicity is potentiated by factors that displace bound bilirubin or otherwise increase circulating levels of unbound bilirubin, such as decreased albumin concentration, free heme molecules, acidosis, increased levels of free fatty acids, or drugs such as sulfonamides and sodium benzoate.1 Infants with severe unconjugated hyperbilirubinemia may develop nerve deafness or the spectrum of kernicterus, leading to severe brain damage, mental retardation, spastic choreoathetosis, or, in many cases, death.
CLINICAL FEATURES
The clinical spectrum of HDFN ranges from the ominous intrauterine development of hydrops fetalis to varying degrees of neonatal hyperbilirubinemia and anemia. Approximately one half of infants with detectable maternal anti-D are unaffected or only mildly affected, whereas 30% have moderate disease in the neonatal period, and approximately 20% are severely affected in utero (Table 30.4).1, 56 The onset of intrauterine disease occurs before 34 weeks’ gestation in approximately one half of the cohort of severely affected fetuses, or approximately 9% of affected pregnancies, overall.56 Similar trends are observed when anti-c, anti-Kl, and anti-Fya are detected in pregnancy, in that many fetuses are unaffected, and most of the rest have only mild or moderate disease, but a small number have hydrops fetalis or severe anemia necessitating intrauterine transfusion (Table 30.4).31, 32, 33, 48, 56, 57 Maternal IgG antibodies with other specificities have been implicated in cases of severe fetal anemia or life-threatening HDN in rare cases.31, 32, 33, 57, 58, 59
Severely affected newborn infants have cord blood hemoglobin concentrations <12 g/dl and cord bilirubin concentrations >5 mg/dl. If intrauterine transfusions have been given, the course of hemolytic disease in the neonatal period may be relatively mild and the blood type at birth may reflect the ABO and D-negative type of the transfused red blood cells. Infants transfused before birth are at risk for developing late anemia at several weeks of age because intrauterine transfusion may suppress erythropoiesis and persistent circulating maternal alloantibody may cause ongoing hemolysis for 4 to 8 weeks. Infants with moderate disease have 12 to 14 g/dl hemoglobin in cord blood and 7 to 12 g/dl hemoglobin in the first days of life. The bilirubin level in cord blood rarely exceeds 5 mg/dl due to maternal clearance. After birth, however, jaundice may occur within the first 24 to 36 hours of life, earlier than the “physiologic jaundice” otherwise associated with immature liver function, and the bilirubin concentration peaks between 3 and 5 days of life. Mild HDN is characterized by cord blood hemoglobin of 14 g/dl or greater and only slightly increased bilirubin (<4 mg/dl).
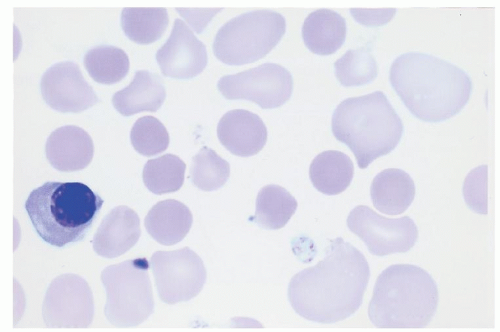
The most common cause of HDN is maternal ABO antibodies, although only a minority of infants with detectable maternal ABO antibodies have clinical signs of hemolysis. Exceedingly rare cases of fetal anemia due to ABO antibodies have been reported, but nonimmune hydrops superimposed on ABO incompatibility could not be excluded.1 Nonimmune hydrops fetalis may be caused by intrauterine infection, cardiac disease, or chromosomal disorders (Table 30.5). Because the neonatal anemia is usually mild, the compensatory hematopoietic activity in ABO HDN is not as pronounced as in HDN due to anti-D or anti-K1. Consequently, nucleated red blood cells and erythropoietic progenitors may be evident, but spherocytes predominate in the peripheral blood smear of infants with ABO HDN (Fig. 30.1). The failure to recognize hemolysis due to ABO blood group incompatibility or other risk factors for hyperbilirubinemia resulted in 90 cases of kernicterus in a 17-year period.60 This contemporary tragedy underscores the ongoing importance of monitoring progressive jaundice in all infants.
TABLE 30.4 CLINICAL SERIES OF RHESUS, KELL, AND DUFFY ALLOANTIBODIES IN PREGNANCY
Severity (% Affected Pregnancies Resulting in Mild, Moderate, or Severe Disease)
Hepatic disease and anatomic abnormalities (e.g., biliary atresia)
Cholestasis from total parenteral nutrition or antibiotics (e.g., ceftriaxone)
LABORATORY EVALUATION
All pregnant women should have their ABO/Rh type and antibody screen determined at the first visit to the obstetrician.61 These initial tests identify women as candidates for RhIG administration or for additional monitoring during pregnancy. After detection of clinically significant red blood cell alloantibodies, further testing may be required for evaluation of FMH, antenatal assessment of fetal anemia, and detection of neonatal anemia and hyperbilirubinemia. The principles of these laboratory tests and controversies surrounding their use are described below; specific practice recommendations for prevention and treatment of HDFN are described in the subsequent sections entitled “Prevention of Maternal D Alloimmunization with Rh Immune Globulin,” “Management of Red Blood Cell Alloimmunization in Pregnancy,” and “Treatment of the Newborn Infant.”
The red blood cell ABO/Rh type is assigned in the forward group reaction by incubating the patient’s red blood cells with specific IgM or IgG antibodies that cause their immediate and visible agglutination if they express the corresponding A, B, or D antigens. The ABO reverse group identifies plasma reactivity due to ABO isoagglutinins and should confirm the forward, red blood cell typing reactions. The forward reaction for the D antigen may include an additional phase with antihuman globulin to enhance red blood cell agglutination and detect weak expression of the D antigen on the red blood cells.
Formerly known as Du, weak D phenotypes are due to quantitative or qualitative alterations in D antigen expression. The need to test D-negative pregnant women for weak expression of D has been controversial. Weak D occurs in as much as 1% to 3% of the general population, but the majority (90%) of weak D individuals (types 1, 2, 3) possess a normal D antigen, only produced in lower quantities on the red blood cell surface, and cannot be immunized to make anti-D.24, 62, 63 The remaining 10% of weak D individuals express aberrant D proteins, and are recognized as partial D phenotypes, many of whom will develop anti-D after exposure to D-positive red blood cells.63, 64 Most D-positive individuals with anti-D belong to partial D category DVI, which occurs in 0.02% to 0.05% of Caucasians.63 The AABB (American Association of Blood Banks) considers testing pregnant women for weak D to be optional, and the American College of Obstetricians and Gynecologists (ACOG) recommends against administration of RhIG to weak-D-positive women.38, 61
Although severe HDF in women with weak D is rare, women with known partial D phenotypes (e.g., DVI) are at risk of D alloimmunization, and fatal HDF has occurred as a result.65, 66 If pregnant women are tested for weak D, consideration should be given to using a method that accurately distinguishes between those individuals with D variants who are not at risk of anti-D alloimmunization from those who are at risk and may benefit from RhIG prophylaxis.63, 64 The alternative is not to test pregnant women (or transfusion recipients) for weak D, which would result in these individuals being classified as D-negative and candidates for RhIG prophylaxis. This presents a challenge to countries that do not have a sufficient supply of RhIG, and not surprisingly, in a recent survey, international practices on testing for weak D and RhIG administration diverge from practice in the United States.40 Eight of ten countries reported that they perform further testing for weak D or D variants if the woman types as D negative or if the typing results are anomalous, to limit administration of RhIG to women with partial D at risk of developing anti-D.40 The relative effectiveness of RhIG in preventing the sensitization of partial D women compared to D-negative women is not yet known.
The antibody screen detects maternal antibody in plasma by hemagglutination in an indirect antiglobulin test, formerly called the indirect Coombs’ test. The reaction conditions should allow detection of clinically significant IgG alloantibodies that are reactive at 37°C in the antiglobulin phase. Conditions that permit identification of IgM antibodies are not required in the setting of obstetric testing, because IgM antibodies cannot cross the placenta. If IgM antibodies cause interference in the antibody screen, reducing agents such as dithiothreitol can be used to eliminate their reactivity in the assay and allow for specific detection of IgG antibodies. If the initial antibody screen is negative, the need to repeat testing at 28 weeks’ gestation is currently debatable. Arguments for eliminating this testing include the extremely low probability that anti-D or other antibodies are formed during pregnancy.46, 67
Serologic and Molecular Testing for Paternal/Fetal Blood Group Antigens
If a potentially significant red blood cell alloantibody is detected in a pregnant woman, the blood type of the biologic father may be investigated to assess the risk of HDFN. Three possibilities exist: the father lacks the antigen, and the fetus is not at risk; the father is heterozygous for the antigen, and the fetus may be at risk; or the father is homozygous for the antigen, and the fetus is definitely at risk. Because no antithetic allele for RhD exists, paternal zygosity cannot be definitively determined by serologic testing for the D antigen but RH D inheritance is closely linked to Rh antigens C/c and E/e. The probability that the father is heterozygous for D can be deduced by serologic determination of the extended Rhesus phenotype (D, Cc, Ee) and most probable combination of haplotypes given the gene frequencies in different ethnic populations and the Rh type of previous children.68 Another indirect approach to determine paternal zygosity involves parallel quantitative amplification of RHD– and RHCE-specific sequence.69 The development of direct, D-allele-specific molecular assays is complicated by the genetic diversity of the D locus and the variety of insertions, deletions, and missense and nonsense mutations in the RHD gene that can produce the D-negative phenotype. Recently, a molecular test specifically to detect the most prevalent D-negative allele in white populations, the complete RHD deletion, was developed.70 This approach may be amenable to routine use in most laboratories, but its clinical application will be limited until modification allows for detection of other common D-negative alleles in ethnically and racially heterogeneous donor populations. Determining the paternal genotype of other blood group determinants is more straightforward with the currently available allele-specific assays for other Rh (C/c, E/e), Kell (K1/K2), Duffy (Fya/Fyb), Kidd (Jka/Jkb), and other red blood cell loci.47, 69
If paternal testing indicates that the father may carry a clinically significant red blood cell antigen, fetal testing should be performed to determine if the allele is present. Considerable genetic diversity underlies aberrant RH alleles associated with the Rh-negative phenotype, which has important implications for fetal testing.47, 69 The most prevalent D-negative genotype in Caucasian populations is the complete deletion of RHD; in African blacks and African Americans, other variant RHD genes are more likely to be found.32 The RHD pseudogene, which contains all ten exons but a stop codon between exons 3 and 4 that blocks transcription of the gene, is found in 69% of South African blacks and 24% of African Americans.32 Similarly, the variant RHD gene r’s (Cdes) underlies serologic RhD negativity in 22% of African Americans. The presence of one of these genes in the fetus can lead to a false-positive result (i.e., the fetus types as RHD-positive by molecular methods but is found to be D negative by serology after birth) and unnecessary prenatal intervention. A maternal blood sample should be analyzed in parallel with the fetal sample. False-negative results (i.e., the fetus types as RHD negative, but is found to be D positive by serology at birth) have been attributed to erroneous paternity or rearrangement at the paternal RHD gene locus. If no paternal sample is available, an RHD-negative fetal blood type determined by molecular methods should be interpreted with caution, and the pregnancy should be monitored to ensure that maternal anti-D titers do not increase.32
Only gold members can continue reading. Log In or Register to continue